Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmelo Massimiliano Rao | -- | 3189 | 2023-09-26 18:16:30 | | | |
| 2 | Rita Xu | Meta information modification | 3189 | 2023-09-27 05:00:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lucà, F.; Oliva, F.; Abrignani, M.G.; Di Fusco, S.A.; Parrini, I.; Canale, M.L.; Giubilato, S.; Cornara, S.; Nesti, M.; Rao, C.M.; et al. Direct Oral Anticoagulants. Encyclopedia. Available online: https://encyclopedia.pub/entry/49684 (accessed on 25 July 2026).
Lucà F, Oliva F, Abrignani MG, Di Fusco SA, Parrini I, Canale ML, et al. Direct Oral Anticoagulants. Encyclopedia. Available at: https://encyclopedia.pub/entry/49684. Accessed July 25, 2026.
Lucà, Fabiana, Fabrizio Oliva, Maurizio Giuseppe Abrignani, Stefania Angela Di Fusco, Iris Parrini, Maria Laura Canale, Simona Giubilato, Stefano Cornara, Martina Nesti, Carmelo Massimiliano Rao, et al. "Direct Oral Anticoagulants" Encyclopedia, https://encyclopedia.pub/entry/49684 (accessed July 25, 2026).
Lucà, F., Oliva, F., Abrignani, M.G., Di Fusco, S.A., Parrini, I., Canale, M.L., Giubilato, S., Cornara, S., Nesti, M., Rao, C.M., Pozzi, A., Binaghi, G., Maloberti, A., Ceravolo, R., Bisceglia, I., Rossini, R., Temporelli, P.L., Amico, A.F., Calvanese, R., ...Gulizia, M.M.. (2023, September 26). Direct Oral Anticoagulants. In Encyclopedia. https://encyclopedia.pub/entry/49684
Lucà, Fabiana, et al. "Direct Oral Anticoagulants." Encyclopedia. Web. 26 September, 2023.
Copy Citation
It is well established that direct oral anticoagulants (DOACs) are the cornerstone of anticoagulant strategy in atrial fibrillation (AF) and venous thromboembolism (VTE) and should be preferred over vitamin K antagonists (VKAs) since they are superior or non-inferior to VKAs in reducing thromboembolic risk and are associated with a lower risk of intracranial hemorrhage (IH).
direct oral anticoagulants (DOACs)
atrial fibrillation (AF)
vitamin K antagonists (VKAs)
adherence
cancer
1. Introduction
Direct oral anticoagulants (DOACs) are currently considered the anticoagulation strategy of choice for thromboembolic prevention in several cardiovascular conditions, such as atrial fibrillation (AF) and venous thromboembolism (VTE) [1][2], and should be preferred over other treatments in eligible patients [3][4].
Indeed, DOACs, classified as direct oral factor Xa inhibitors (apixaban, rivaroxaban, and edoxaban) and direct thrombin inhibitors (dabigatran), have been demonstrated to be superior or non-inferior to vitamin K antagonists (VKAs) in lowering the risk of thromboembolic events without increasing bleeding risk [2][5]. Moreover, many advantages have been reported, including the fact that they do not need coagulation parameters, have fewer nutrient and drug–drug interactions (DDIs), and present fewer other pharmacokinetic and pharmacodynamic issues, such as genetic polymorphisms, which may affect VKAs’ activity [2].
However, several aspects of DOAC management remain challenging, including DDIs, switching to other therapies, periprocedural and postprocedural management strategies, and the use in patients with chronic renal and liver failure and in those with cancer [2]. Moreover, according to real-world data, adherence to DOACs seems to be scarce, remaining at approximately 40% of DOAC patients [6].
2. Drug–Drug Interactions (DDIs) of DOACs and Polypharmacy
Although fewer DDIs have been ascribed to DOAC than expected using warfarin [7], the concomitant administration of agents that affect DOAC plasma concentrations may increase or reduce DOAC effects, potentially increasing bleeding or ischemic risk, respectively.
The principal interactions of DOACs that need to be considered concern drugs that influence renal and hepatic clearance and drugs that affect hemostasis [8]. The interactions with the efflux transporter P-glycoprotein (P-gp) and CYP3A4-type cytochrome P450 (CYP3A4/5) must also be carefully taken into account [7].
From the perspective of DDI, medications can be categorized as inducers or inhibitors of one or more of these enzymes or transport proteins. In cases of inhibition, there arises direct competition between medications, leading to heightened serum concentrations of either one or both agents. Conversely, induction results in diminished serum concentrations, potentially compromising the effectiveness of a medication [9][10].
With regard to CYP450 enzymes, those most commonly involved in DDI are CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5 [9][11].
It has been shown that hepatic clearance of both rivaroxaban and apixaban is CYP3A4 type cytochrome P450-dependent [12][13], considering the fact that CYP3A4 contributes to approximately 50% and 20–25% of their respective metabolic pathways [10]. This differs from dabigatran, which does not act as a substrate, inhibitor, or inducer of Cytochrome P450 enzymes, and edoxaban, where less than 4% of its metabolism is facilitated by the CYP3A4 enzyme [9][10].
P-gp belongs to the multidrug resistance protein 1 (MDR1) family, which is encoded by the ATP-binding cassette subfamily B (ABCB1) gene [9].
Additionally, P-gp operates as an efflux pump, playing a pivotal role in mitigating tissue exposure to compounds with potentially deleterious effects, thereby facilitating their efflux and removal [10][11].
P-GP takes part in the renal excretion of DOACs [8]. Thus, the P-GP competitive inhibition will increase DOAC plasma values [14].
P-gp acts as a substrate for apixaban, dabigatran, and rivaroxaban, while it does not exhibit any significant activity on edoxaban [9].
However, also other transport proteins and enzymes have been hypothesized to be involved in DDIs, such as the influx transporter, the organic anion-transporter polyprotein (OATP), the efflux/influx organic transporter (OCT), and the efflux transporter breast cancer resistance protein (BCRP), expressed in the intestinal, hepatic, and biliary sites [9]. This fact may represent a problem because AF or VTE patients are commonly politreated. By and large, polymedication has been correlated with increased mortality and bleeding [15][16][17].
The effects of DDIs have been extensively described in the ESC practical guide on DOACs [18]. Strong CYP3A4/5 and P-GP/CYP3A4/5 inhibitors can significantly increase DOAC plasma levels and increase the risk of bleeding [19][20]. If strong CYP3A4/5 inhibitors need to be used, it is preferable to use dabigatran, edoxaban, or VKAs rather than apixaban and rivaroxaban, considering that CYP3A4/5 affects their metabolism more. Of note, only for edoxaban, a dose reduction is recommended in patients concomitantly taking potent P-GP inhibitors, including dronedarone, verapamil, and quinidine, according to the ENGAGE study [21]. Since all DOACs are substrates for PGP, strong PGP/CYP 3A4/5 inhibitors expose patients to increased bleeding risk with all DOACs [7]. Antifungals, macrolides, and antiretroviral protease inhibitors are potent inhibitors of PGP that interact with DOACs [7].
DOACs may be cautiously used concomitantly with moderate and weak inhibitors of PGP or CYP 3A4/5, providing that other bleeding risk factors such as renal dysfunction (clearance < 50 mL/min), weight < 60 kg, or advanced age (>80 years) did not coexist. In addition, for rivaroxaban, the presence of a mild or moderate hepatic failure (Child-Pugh A or B) represents a condition that requires careful management if moderate-weak inhibitors of PGP or CYP 3A4/5 have to be contemporarily used. Moreover, when a moderate- weak PGP or CYP 3A4/5 inhibitor must be associated with DOACs, if more than two bleeding risk factors occur, or if a severe renal dysfunction coexists (clearance < 30 mL/min), it should be reasonable to use VKAs as a first-line treatment or, if it is possible, to choose a non-interacting drug.
The assessment of DOAC plasmatic levels and the use of “off-label” reduced doses are not supported by consistent evidence, so this strategy is not suggested for most patients. In particular, the determination of DOAC plasma concentrations should be limited to rare cases when potentially significant interactions occur or for specific conditions as an option only for experienced centers.
On the other hand, moderate-strong PGP/CYP3A4/5 inducers’ use should be avoided in all patients taking DOACs as they determine a significant reduction in DOAC concentration. Therefore, VKAs may be considered the best option in these cases.
For example, DOACs administration is contraindicated in patients treated with rifampicin, a potent inducer of PGP. Rivaroxaban and apixaban labels advise avoiding the use of carbamazepine, phenytoin, phenobarbital, concomitant P-GP, and CYP3A4 inducers.
A list of the most common drugs, moderate to strong Pgp/CYP3A4/5 inhibitors, and inducers is shown in Table 1.
Table 1. Drug–Drug Interactions (DDIs) of DOACs.
| Drugs | Strong | Moderate to Weak |
|---|---|---|
| Pgp or combined CYP3A4/5/Pgp inhibitors | Clarithromycin, Cobicistat, Ketoconazole, Itraconazole, Dronedarone, Erythromycin, Posaconazole, Ritonavir, Voriconazole | Amiodarone, Cyclosporine, Diltiazem, Ticagrelor, Verapamil, Quinidine |
| CYP3A4/5 inhibitors | Boceprevir, Grapefruit Juice | Fluconazole |
| P-gp inducers | Rifampin | |
| CYP3A4/5 inducers | Phenytoin | |
| CYP3A4/5 inducer+combined P-gp inducer | Apalutamide, Bosentan, Carbamazepine, Phenobarbital, St. John’s Wort |
Pgp: P-glycoprotein; CYP3A4/5: cytochrome P450 3A4/5.
Although DOACs are recognized for their predictable pharmacokinetics and pharmacodynamics, recent observations have drawn attention to a wider spectrum of inter-individual variability encompassing both plasma concentrations and drug responses. This variability can be influenced by several factors, including age, race, gender, smoking, and dietary patterns. Moreover, the presence of common genetic variations or interactions between drugs may also contribute to the manifestation of these differences [22].
The exploration of pharmacogenomics in relation to DOACs constitutes a relatively nascent field of investigation. However, it should place more emphasis on personalized medication management and pharmacogenomic testing to optimize DOAC prescribing in patients with potential interactions. Moreover, it is advisable to gain insight into the contribution of pharmacogenomics to the interpatient diversity observed in DOAC responses. Indeed, the variability exhibited in DOAC responses can be partially ascribed to genetic variants within specific gene loci as well as drug–drug interactions [22].
Remarkably, genetic variants within carboxylesterase 1 (CES1) and ABCB1 with multiple single nucleotide polymorphisms (SNPs) are among the most extensively documented, contributing to notable modifications in the peak and trough levels of dabigatran, yielding discernible clinical ramifications. Similarly, ABCB1 SNPs exert an influence on the modulation of plasma drug concentrations of rivaroxaban and apixaban. Conversely, investigations involving genetic variants such as factor Xa, ABCB1, Solute Carrier Organic Anion Transporter Family Member 1B1 (SLCOB1), CYP2C9, and Vitamin K epOxide Reductase Complex (VKORC1) subunit 1 did not uncover any substantial associations with the plasma drug levels of edoxaban [22].
3. How to Switch between Different Anticoagulants
Patients on anticoagulation therapy may need a switch from DOACs to VKAs or vice versa for several reasons [23]. In these cases, it is of paramount importance to consider the different pharmacokinetic and pharmacodynamics proprieties among different anticoagulants, ensuring the continuation of therapy and reducing the bleeding risk.
VKAs to DOACs. According to the latest international recommendations, an INR < 2 allows the immediate initiation of DOACs, whereas, in the presence of INR 2.0–2.5, DOACs could be administrated promptly or postponed to the next day [24]. If the patient has an INR value above 2.5, it is advisable to take into account the time needed for obtaining an INR lower than the threshold value, considering a half time of 8–24 h for acenocoumarol, 36–48 h for warfarin, and six days for phenprocoumon [24]. At this time, a second INR measurement is recommended before DOAC administration. When DOACs are started, no further INR measurements are needed [24].
DOACs to VKAs. Due to the slow time to achieve the therapeutic range of VKAs, from 5–10 days, a simultaneous administration of DOACs and VKAs is recommended until INR reaches an appropriate therapeutic value [24]. No loading dose is recommended for VKAs, but for phenprocoumon. Notably, since DOACs may affect INR, this should be closely monitored for at least one month after DOACs withdraw, until stable INR values are achieved. Particularly, 2–3 days after stopping DOACs, a new measurement is strongly suggested to ensure adequate anticoagulation.
If concurrent DOAC administration during VKA treatment initiation is considered inappropriate (i.e., severe acute renal impairment with secondary increased DOAC blood levels), switching from DOACs to Low-Molecular-Weight Heparin (LMWH) with concomitant VKAs administration may be a suitable option, chiefly in high thromboembolic risk patients [24].
DOACs to DOACs. The alternative DOACs should be administrated at the time the next dose of the initial DOACs is expected. It has been suggested that a longer interval is seen in the presence of situations in which plasma concentrations are expected to be higher than the therapeutic concentration (i.e., renal impairment). However, no further and more detailed recommendations based on time administration have been provided.
DOACs to parenteral or subcutaneous anticoagulation. Parenteral or subcutaneous anticoagulation treatment should be started when the next DOAC dose is due [24]. However, if ST-elevation myocardial infarction (STEMI) occurs, enoxaparin or unfractionated heparin (UFH) should be used regardless of the time of the last dose of DOACs [24].
Parenteral anticoagulant to DOACs. Due to a very short half-life of intravenous UFH (2 h), DOACs can be started 4 h after intravenous UFH discontinuation [24]. Conversely, in the presence of LMWH treatment, DOACs should be administrated when the next LMWH dose is due, with particular attention if a renal dysfunction coexists wherein LMWH catabolism may be longer.
DOACs in patients with Chronic kidney disease (CKD)
Chronic kidney disease (CKD) and AF may increase thromboembolic risk through several mechanisms [25]. Moreover, the bleeding risk has been shown to rise in CKD. The risk of bleeding is particularly high in the presence of GFR < 30 mL/m, CKD-related anemia, polytherapy, and invasive procedures.
The gradual development of renal impairment has been estimated to increase the risk of cerebral hemorrhage up to 10-fold in dialysis patients [26].
In addition, GI bleeding has been reported to increase with worsening renal function [27].
For this reason, managing AF patients with concomitant CKD is particularly challenging.
Patient risk stratification should be performed using the CHA2DS2VASc and HAS-BLED scores. Before starting oral anticoagulant therapy, all measures to lower bleeding risk must be implemented. All DOACs are eliminated by the kidney; renal excretion of 80%, 50%, 35%, and 27% has been observed for dabigatran edoxaban, rivaroxaban, and apixaban, respectively.
Therefore, before administering DOACs, an evaluation of renal function is required [28].
Any condition that may worsen renal function (infection, acute heart failure, potentially nephrotoxic drugs, etc.) requires additional checks. Obviously, DOACs should be suspended for patients with acute renal failure [29].
The dosage to use in patients with mild-moderate CKD, defined as a glomerular filtration rate (GFR) between 30 and 50 mL/min, depends on each DOAC’s kidney elimination. In these patients, dabigatran, 110/150 mg can be used according to the patient’s clinical characteristics, apixaban 2.5/5 mg should be used following specific parameters extrapolated from phase 3 clinical trials, and the dosage of rivaroxaban must be reduced to 15 mg if creatinine serum levels are >1.5 mg/dL and edoxaban needs to be reduced at 30 mg when GFR is between 15 and 49 mL/min [18]. In the US and Europe, the use of low doses of rivaroxaban, apixaban, and edoxaban (but not dabigatran) has been approved with a CrCl of 15–29 mL/min [18], although data on outcomes are poor.
According to the guidelines for GFR < 15 mL/min, DOACs are contraindicated considering the fact that phase 3 clinical trials did not include dialytic and advanced renal dysfunction individuals [18].
Moreover, limited studies on apixaban and rivaroxaban in patients with end-stage renal disease (ESRD) and/or receiving hemodialysis hypothesize safety and efficacy findings in this complex category [30][31][32].
However, whether patients with severe renal impairment may benefit from DOACs and, furthermore, which drug should be used in this case, has yet to be substantially confirmed. When a patient is on dialysis or has a GFR < 15 mL/min, guidelines suggest an individualized approach that includes a multidisciplinary team discussion and patient engagement with a shared decision approach after being informed of the off-label use of drugs [29][32][33].
Another point regarding DOACs and CKD that deserves to be mentioned is the risk of CKD progression determined by anticoagulant treatments. Indeed, anticoagulant-related nephropathy is a rare disease determined by renovascular calcification and intrarenal hemorrhages. This condition can affect patients using both warfarin and DOACs, presenting as an acute kidney injury of a progressive CKD cell cast in renal tubules [33][34].
4. DOACs in Patients with Advanced Chronic Liver Disease (CLD)
Normal hepatic function is of paramount importance to balance homeostasis and anti-thrombotic function [35][36][37] (Figure 1). In this regard, people with advanced chronic liver disease (CLD) run a greater risk of both thrombosis and bleeding risk, which is related to thrombocytopenia (secondary to reduced thrombopoietin production) and reduced synthesis of fibrinogen or other coagulation factors such as II, V, VII, IX, X, XI, and XII. Conversely, low levels of protein C, antithrombin, and plasminogen, and, at the same time, hypercoagulability due to enhanced von Willebrand factor activity imply increased thrombotic risk. Of note, advanced CLD is commonly associated with prolonged prothrombin time [38][39].
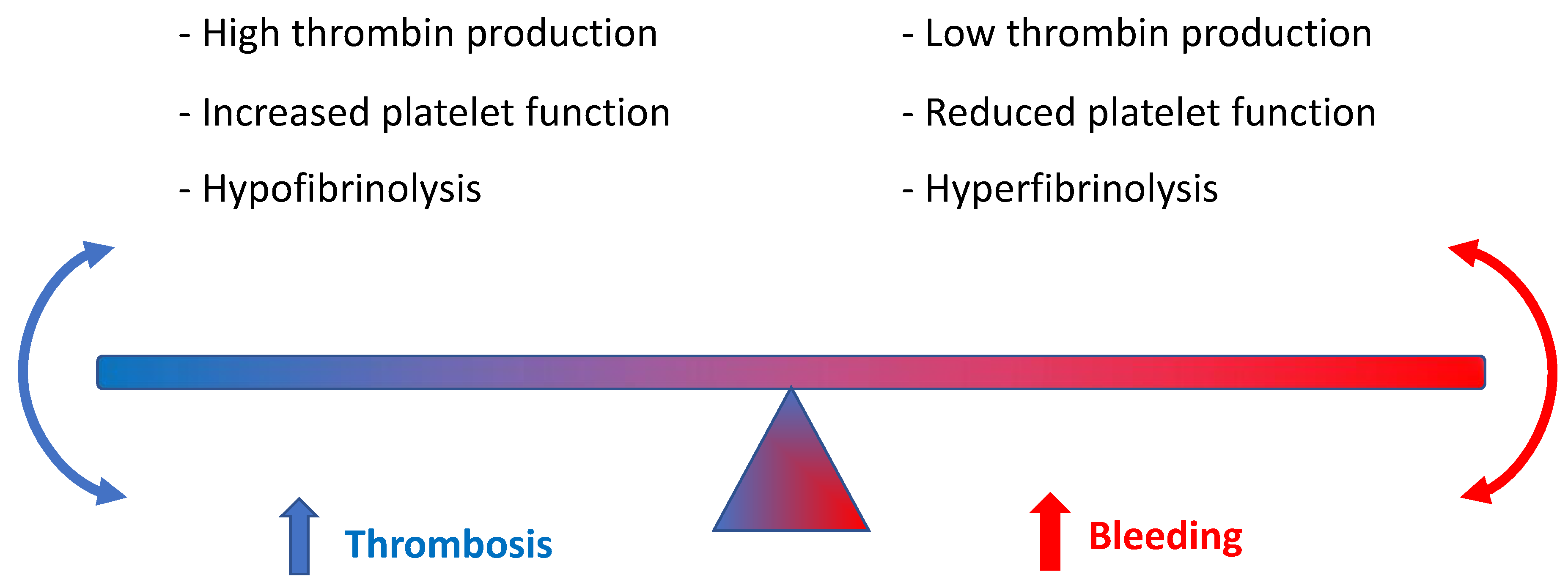
Figure 1. Role of hepatic function in balancing homeostasis and anti-thrombotic function.
The aforementioned characteristics represent a great challenge in managing OAC therapy when AF or VTE are associated with CLD. Moreover, data concerning DOACs in patients with CLD are poor since they were scarcely represented in DOAC trials [21][40][41][42].
However, the effect and drug concentration of DOACs may be affected by the presence of CLD considering the fact that hepatic metabolism of 75%, 65%, and 50% has been reported for apixaban, rivaroxaban, and edoxaban, respectively [18]. Conversely, dabigatran etexilate is a prodrug and its metabolism is not affected by hepatic function.
The European Medicines Agency (EMA) and the Food and Drug Administration (FDA) consider the Child–Pugh classification to guide DOACs use in patients with CLD. The Child–Pugh classification is a score contemplating clinical (ascites and encephalopathy) and serum values such as albumin, bilirubin, prothrombin time, or INR parameters to evaluate the prognosis of patients with CLD. The Child–Pugh classification classifies patients with mild (class A), moderate (class B), and severe (class C) liver impairment [18].
According to the FDA label recommendation, apixaban does not need any dose adjustment in patients with Child–Pugh A. As for Child–Pugh B patients, its use has been suggested with caution in consideration of scarce data, even though dose adjustment is not needed. Conversely, in Child–Pugh C patients, apixaban use is not recommended [18]. Notably, liver function should be assessed before administering apixaban in patients with CLD due to the fact that if a concomitant coagulation disorder and clinically relevant bleeding risk coexist, its use should be avoided [18].
As previously reported, two-thirds of rivaroxaban have hepatic metabolism. According to the pharmacokinetic and pharmacodynamic properties, rivaroxaban should be avoided in Child–Pugh B and C classes or in the presence of a concomitant coagulopathy.
Due to the high hepatic metabolism of edoxaban, any dose adjustment in Child–Pugh A patients is required. In contrast, this Xa inhibitor is not recommended in Child–Pugh B and C adults. Conversely, according to the EMA recommendation, edoxaban does not need any dose adjustment in Child–Pugh A and B patients, whereas in Child–Pugh C patients, it is not recommended. Additionally, according to EMA, these Xa inhibitors should not be used in CLD patients with coagulation disorders and in those at high bleeding risk [18].
Dabigatran etexilate is the sole prodrug among DOACs, and due to a reduced metabolized fraction in the liver, hepatic impairment has less influence on its metabolism. According to DOACs’ pharmacokinetics and pharmacodynamics, patients with mild or moderate CLD should not have a dose adjustment, whereas, in the Child–Pugh C population, dabigatran is not recommended [18]. Conversely, the use of dabigatran should be avoided in patients with a 2-fold increased upper limit of hepatic enzyme regardless of hepatic impairment, and the oral thrombin inhibitor is not recommended in patients with advanced CLD.
As previously reported, patients with CLD run a higher bleeding risk because of the reduced production of pro-thrombotic agents. [18].
Differently from patients with renal impairment, there is a paucity of information regarding DOAC use in patients suffering from liver disorders.
Indeed, the CLD population has been largely excluded from pivotal randomized controlled trials investigating antithrombotic medications. This has led to a poor agreement regarding the safety, effectiveness, and monitoring protocols associated with anticoagulant and antiplatelet therapies in patients with CLD. As a consequence, the optimal strategy for DOAC in the CLD patient cohort remains an area of uncertainty.
Importantly, to date, no prospective clinical trial has examined the safety and efficacy of DOACs in reducing thrombotic events in patients with CLD, and available data have been obtained from pharmacokinetic investigations, case reports, and limited-scale observational research [43][44][45][46].
Furthermore, managing CLD patients in terms of anticoagulation therapy is particularly challenging in high-risk contexts such as acute coronary syndrome (ACS) and urgent percutaneous revascularization (PCI). Indeed, altered hemostatic mechanisms have been associated with liver impairment so OAC use combined with one or more antiplatelet therapies should be restricted to the shortest period in this high-risk population.
Considering the growing incidence of AF and CAD in this subset of patients and the expanding range of therapeutic options, data were extrapolated from real-world analyses [43].
DOAC-treated patients with advanced CLD and bleeding episodes should be considered as those without hepatic dysfunction. Thrombin time, diluted thrombin time, and ecarin clot time are the coagulation tests used in patients on dabigatran. In contrast, a calibrated chromogenic anti-Xa assay is commonly used in patients treated with Xa inhibitors [47][48][49]. These tests assess the blood concentration of DOACs. If the concentration exceeds 30 ng/mL, reversal agents such as Idaracuzimab and Andexanet alfa for dabigatran and Xa inhibitor, respectively, should be considered [18][50][51].
References
- Aronis, K.N.; Hylek, E.M. Evidence gaps in the era of Non–Vitamin K oral anticoagulants. J. Am. Heart Assoc. 2018, 7, e007338.
- Rose, D.K.; Bar, B. Direct oral anticoagulant agents: Pharmacologic profile, indications, coagulation monitoring, and reversal agents. J. Stroke Cerebrovasc. Dis. 2018, 27, 2049–2058.
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Furie, K.L. 2019 AHA/ACC/HRS focused update of the 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society in collaboration with the Society of Thoracic Surgeons. Circulation 2019, 140, e125–e151.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498.
- Kapoor, A.; Ellis, A.; Shaffer, N.; Gurwitz, J.; Chandramohan, A.; Saulino, J.; Ishak, A.; Okubanjo, T.; Michota, F.; Hylek, E. Comparative effectiveness of venous thromboembolism prophylaxis options for the patient undergoing total hip and knee replacement: A network meta-analysis. J. Thromb. Haemost. 2017, 15, 284–294.
- Salmasi, S.; Loewen, P.S.; Tandun, R.; Andrade, J.G.; De Vera, M.A. Adherence to oral anticoagulants among patients with atrial fibrillation: A systematic review and meta-analysis of observational studies. BMJ Open 2020, 10, e034778.
- Mar, P.L.; Gopinathannair, R.; Gengler, B.E.; Chung, M.K.; Perez, A.; Dukes, J.; Ezekowitz, M.D.; Lakkireddy, D.; Lip, G.Y.H.; Miletello, M.; et al. Drug Interactions Affecting Oral Anticoagulant Use. Circ. Arrhythm. Electrophysiol. 2022, 15, e007956.
- Chen, A.; Stecker, E.; Warden, B.A. Direct Oral Anticoagulant Use: A Practical Guide to Common Clinical Challenges. J. Am. Heart Assoc. 2020, 9, e017559.
- Wiggins, B.S.; Dixon, D.L.; Neyens, R.R.; Page, R.L., 2nd; Gluckman, T.J. Select Drug-Drug Interactions With Direct Oral Anticoagulants: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 1341–1350.
- Burnett, A.E.; Mahan, C.E.; Vazquez, S.R.; Oertel, L.B.; Garcia, D.A.; Ansell, J. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J. Thromb. Thrombolysis 2016, 41, 206–232.
- Vazquez, S.R. Drug-drug interactions in an era of multiple anticoagulants: A focus on clinically relevant drug interactions. Blood 2018, 132, 2230–2239.
- Mueck, W.; Kubitza, D.; Becka, M. Co-administration of rivaroxaban with drugs that share its elimination pathways: Pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 455–466.
- Heidbuchel, H.; Verhamme, P.; Alings, M.; Antz, M.; Hacke, W.; Oldgren, J.; Sinnaeve, P.; Camm, A.J.; Kirchhof, P. European Heart Rhythm Association Practical Guide on the use of new oral anticoagulants in patients with non-valvular atrial fibrillation. Europace 2013, 15, 625–651.
- Steffel, J.; Verhamme, P.; Potpara, T.S.; Albaladejo, P.; Antz, M.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; et al. The 2018 European Heart Rhythm Association Practical Guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur. Heart J. 2018, 39, 1330–1393.
- Nobili, A.; Marengoni, A.; Tettamanti, M.; Salerno, F.; Pasina, L.; Franchi, C.; Iorio, A.; Marcucci, M.; Corrao, S.; Licata, G.; et al. Association between clusters of diseases and polypharmacy in hospitalized elderly patients: Results from the REPOSI study. Eur. J. Intern. Med. 2011, 22, 597–602.
- Proietti, M.; Raparelli, V.; Olshansky, B.; Lip, G.Y. Polypharmacy and major adverse events in atrial fibrillation: Observations from the AFFIRM trial. Clin. Res. Cardiol. 2016, 105, 412–420.
- Leiss, W.; Méan, M.; Limacher, A.; Righini, M.; Jaeger, K.; Beer, H.J.; Osterwalder, J.; Frauchiger, B.; Matter, C.M.; Kucher, N.; et al. Polypharmacy is associated with an increased risk of bleeding in elderly patients with venous thromboembolism. J. Gen. Intern. Med. 2015, 30, 17–24.
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. Europace 2021, 23, 1612–1676.
- Herink, M.C.; Zhuo, Y.F.; Williams, C.D.; DeLoughery, T.G. Clinical Management of Pharmacokinetic Drug Interactions with Direct Oral Anticoagulants (DOACs). Drugs 2019, 79, 1625–1634.
- Foerster, K.I.; Hermann, S.; Mikus, G.; Haefeli, W.E. Drug-Drug Interactions with Direct Oral Anticoagulants. Clin. Pharmacokinet. 2020, 59, 967–980.
- Giugliano, R.P.; Ruff, C.T.; Braunwald, E.; Murphy, S.A.; Wiviott, S.D.; Halperin, J.L.; Waldo, A.L.; Ezekowitz, M.D.; Weitz, J.I.; Špinar, J.; et al. Edoxaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2013, 369, 2093–2104.
- Kanuri, S.H.; Kreutz, R.P. Pharmacogenomics of Novel Direct Oral Anticoagulants: Newly Identified Genes and Genetic Variants. J. Pers. Med. 2019, 9, 7.
- Barrett, A.; Moore, M.; Ferrins, P.; Thornton, P.; Murphy, P.; Quinn, J. From a direct oral anticoagulant to warfarin: Reasons why patients switch. Ir. J. Med. Sci. 2018, 187, 719–721.
- Steffel, J.; Heidbüchel, H. 2021 European Heart Rhythm Association Practical Guide on the use of non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation: Comment-Authors’ reply. Europace 2021, 23, 1685–1686.
- Keller, C.; Katz, R.; Cushman, M.; Fried, L.F.; Shlipak, M. Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: A cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA). BMC Nephrol. 2008, 9, 9.
- Iseki, K.; Kinjo, K.; Kimura, Y.; Osawa, A.; Fukiyama, K. Evidence for high risk of cerebral hemorrhage in chronic dialysis patients. Kidney Int. 1993, 44, 1086–1090.
- Sood, P.; Kumar, G.; Nanchal, R.; Sakhuja, A.; Ahmad, S.; Ali, M.; Kumar, N.; Ross, E.A. Chronic kidney disease and end-stage renal disease predict higher risk of mortality in patients with primary upper gastrointestinal bleeding. Am. J. Nephrol. 2012, 35, 216–224.
- Lucà, F.; Giubilato, S.; Di Fusco, S.A.; Piccioni, L.; Rao, C.M.; Iorio, A.; Cipolletta, L.; D’Elia, E.; Gelsomino, S.; Rossini, R.; et al. Anticoagulation in Atrial Fibrillation Cardioversion: What Is Crucial to Take into Account. J. Clin. Med. 2021, 10, 3212.
- Aursulesei, V.; Costache, I.I. Anticoagulation in chronic kidney disease: From guidelines to clinical practice. Clin. Cardiol. 2019, 42, 774–782.
- Ionescu, F.; Cooper, C.; Petrescu, I.; George, J.; Mansuri, S. Safety of apixaban compared to warfarin in hemodialysis patients: Do antiplatelets make a difference? Eur. J. Haematol. 2021, 106, 689–696.
- Ellenbogen, M.I.; Ardeshirrouhanifard, S.; Segal, J.B.; Streiff, M.B.; Deitelzweig, S.B.; Brotman, D.J. Safety and effectiveness of apixaban versus warfarin for acute venous thromboembolism in patients with end-stage kidney disease: A national cohort study. J. Hosp. Med. 2022, 17, 809–818.
- Miao, B.; Sood, N.; Bunz, T.J.; Coleman, C.I. Rivaroxaban versus apixaban in non-valvular atrial fibrillation patients with end-stage renal disease or receiving dialysis. Eur. J. Haematol. 2020, 104, 328–335.
- Kumar, S.; Lim, E.; Covic, A.; Verhamme, P.; Gale, C.P.; Camm, A.J.; Goldsmith, D. Anticoagulation in Concomitant Chronic Kidney Disease and Atrial Fibrillation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2204–2215.
- Chantrarat, T.; Hauythan, S. The change of renal functions after nonvitamin K oral anticoagulants in patients with atrial fibrillation. Int. J. Cardiol. Heart Vasc. 2021, 35, 100844.
- Kujovich, J.L. Coagulopathy in liver disease: A balancing act. Hematology 2015, 2015, 243–249.
- Amitrano, L.; Guardascione, M.A.; Brancaccio, V.; Balzano, A. Coagulation disorders in liver disease. Semin. Liver Dis. 2002, 22, 83–96.
- Northup, P.G.; Caldwell, S.H. Coagulation in Liver Disease: A Guide for the Clinician. Clin. Gastroenterol. Hepatol. 2013, 11, 1064–1074.
- Northup, P.G.; McMahon, M.M.; Ruhl, A.P.; Altschuler, S.E.; Volk-Bednarz, A.; Caldwell, S.H.; Berg, C.L. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am. J. Gastroenterol. 2006, 101, 1524–1528, quiz 1680.
- Dabbagh, O.; Oza, A.; Prakash, S.; Sunna, R.; Saettele, T.M. Coagulopathy does not protect against venous thromboembolism in hospitalized patients with chronic liver disease. Chest 2010, 137, 1145–1149.
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Eikelboom, J.; Oldgren, J.; Parekh, A.; Pogue, J.; Reilly, P.A.; Themeles, E.; Varrone, J.; et al. Dabigatran versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 361, 1139–1151.
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992.
- Patel, M.R.; Mahaffey, K.W.; Garg, J.; Pan, G.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; Hankey, G.J.; Piccini, J.P.; et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 2011, 365, 883–891.
- Chang, W.H.; Mueller, S.H.; Tan, Y.Y.; Lai, A.G. Antithrombotic therapy in patients with liver disease: Population-based insights on variations in prescribing trends, adherence, persistence and impact on stroke and bleeding. Lancet Reg. Health Eur. 2021, 10, 100222.
- Graff, J.; Harder, S. Anticoagulant therapy with the oral direct factor Xa inhibitors rivaroxaban, apixaban and edoxaban and the thrombin inhibitor dabigatran etexilate in patients with hepatic impairment. Clin. Pharmacokinet. 2013, 52, 243–254.
- Qamar, A.; Vaduganathan, M.; Greenberger, N.J.; Giugliano, R.P. Oral Anticoagulation in Patients With Liver Disease. J. Am. Coll. Cardiol. 2018, 71, 2162–2175.
- Lawal, O.D.; Aronow, H.D.; Shobayo, F.; Hume, A.L.; Taveira, T.H.; Matson, K.L.; Zhang, Y.; Wen, X. Comparative Effectiveness and Safety of Direct Oral Anticoagulants and Warfarin in Patients With Atrial Fibrillation and Chronic Liver Disease: A Nationwide Cohort Study. Circulation 2023, 147, 782–794.
- Božič-Mijovski, M.; Malmström, R.E.; Malovrh, P.; Antovic, J.P.; Vene, N.; Šinigoj, P.; Mavri, A. Diluted thrombin time reliably measures low to intermediate plasma dabigatran concentrations. Ann. Clin. Biochem. 2016, 53, 446–451.
- Lange, U.; Nowak, G.; Bucha, E. Ecarin chromogenic assay--a new method for quantitative determination of direct thrombin inhibitors like hirudin. Pathophysiol. Haemost. Thromb. 2003, 33, 184–191.
- Beyer, J.; Trujillo, T.; Fisher, S.; Ko, A.; Lind, S.E.; Kiser, T.H. Evaluation of a Heparin-Calibrated Antifactor Xa Assay for Measuring the Anticoagulant Effect of Oral Direct Xa Inhibitors. Clin. Appl. Thromb. Hemost. 2016, 22, 423–428.
- Connolly, S.J.; Crowther, M.; Eikelboom, J.W.; Gibson, C.M.; Curnutte, J.T.; Lawrence, J.H.; Yue, P.; Bronson, M.D.; Lu, G.; Conley, P.B.; et al. Full Study Report of Andexanet Alfa for Bleeding Associated with Factor Xa Inhibitors. N. Engl. J. Med. 2019, 380, 1326–1335.
- Gottlieb, M.; Khishfe, B. Idarucizumab for the Reversal of Dabigatran. Ann. Emerg. Med. 2017, 69, 554–558.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
692
Revisions:
2 times
(View History)
Update Date:
27 Sep 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No