Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chaiyavat Chaiyasut | -- | 2352 | 2023-09-25 10:38:14 | | | |
| 2 | Camila Xu | Meta information modification | 2352 | 2023-09-25 10:46:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sivamaruthi, B.S.; Raghani, N.; Chorawala, M.; Bhattacharya, S.; Prajapati, B.G.; Elossaily, G.M.; Chaiyasut, C. NF-κB Pathway Inhibitors in Alzheimer’s Disease Treatment. Encyclopedia. Available online: https://encyclopedia.pub/entry/49590 (accessed on 15 June 2026).
Sivamaruthi BS, Raghani N, Chorawala M, Bhattacharya S, Prajapati BG, Elossaily GM, et al. NF-κB Pathway Inhibitors in Alzheimer’s Disease Treatment. Encyclopedia. Available at: https://encyclopedia.pub/entry/49590. Accessed June 15, 2026.
Sivamaruthi, Bhagavathi Sundaram, Neha Raghani, Mehul Chorawala, Sankha Bhattacharya, Bhupendra G. Prajapati, Gehan M. Elossaily, Chaiyavat Chaiyasut. "NF-κB Pathway Inhibitors in Alzheimer’s Disease Treatment" Encyclopedia, https://encyclopedia.pub/entry/49590 (accessed June 15, 2026).
Sivamaruthi, B.S., Raghani, N., Chorawala, M., Bhattacharya, S., Prajapati, B.G., Elossaily, G.M., & Chaiyasut, C. (2023, September 25). NF-κB Pathway Inhibitors in Alzheimer’s Disease Treatment. In Encyclopedia. https://encyclopedia.pub/entry/49590
Sivamaruthi, Bhagavathi Sundaram, et al. "NF-κB Pathway Inhibitors in Alzheimer’s Disease Treatment." Encyclopedia. Web. 25 September, 2023.
Copy Citation
The NF-κB family is a group of transcription factors that play a pivotal role in regulating various biological processes, including immune responses, inflammation, cell survival, and cellular differentiation. The involvement of the NF-κB pathway in immune system responses, inflammation, oxidative stress, and neuronal survival highlights its significance in Alzheimer’s disease (AD) progression.
Alzheimer’s disease
NF-κB
amyloid beta plaques
neuroinflammation
1. Introduction
Alzheimer’s disease (AD), a neurodegenerative disease, is the most common type of dementia, responsible for up to 75% of all cases [1]. It manifests as a clinical syndrome presenting symptoms like impairment of memory, language, and other cognitive functions, and behavioral changes that severely affect the activities of daily living. The worldwide prevalence of dementia is thought to be around 3.9%, which is estimated to escalate with the growing size of the population aged 65 years and older. The fundamental histopathological feature of AD has been considered to be aggregates of amyloid beta (Aβ) plaques in the cortical and limbic regions of the brain [2]. Despite offering a comprehensive framework for understanding AD pathogenesis, certain observations do not seamlessly align with the most straightforward rendition of the hypothesis, leading to its existing lack of intricate explanations [3][4][5]. It is noteworthy that, intriguingly, no treatment closely tied to the proposed amyloid hypothesis has achieved success thus far. Thus, ineffective attempts to manage the progressive symptoms have compelled new insights into pathogenesis. Post-translational modifications (PTMs) are pivotal in AD as they influence the aggregation of pathogenic proteins implicated in the disease [6]. Aberrant PTMs, such as the hyperphosphorylation of tau protein and Aβ, glycation, nitration, and ubiquitination, contribute to the misfolding and aggregation of these proteins. The phosphorylation of tau disrupts its normal function, leading to neurofibrillary tangles (NFTs). Aβ, through PTMs like glycation and nitration, adopts toxic conformations, forming amyloid plaques. Ubiquitination may impair the clearance of misfolded proteins. These PTMs disrupt protein homeostasis, impair cellular functions, and trigger neuroinflammation. The interplay between PTMs and nuclear factor kappa B (NF-κB)-driven inflammation thus contributes to protein aggregation, neurodegeneration, and AD progression.
The NF-κB family is a group of transcription factors that play a pivotal role in regulating various biological processes, including immune responses, inflammation, cell survival, and cellular differentiation [7]. The family encompasses five members. NF-κB1 regulates immune responses and inflammation (Figure 1).
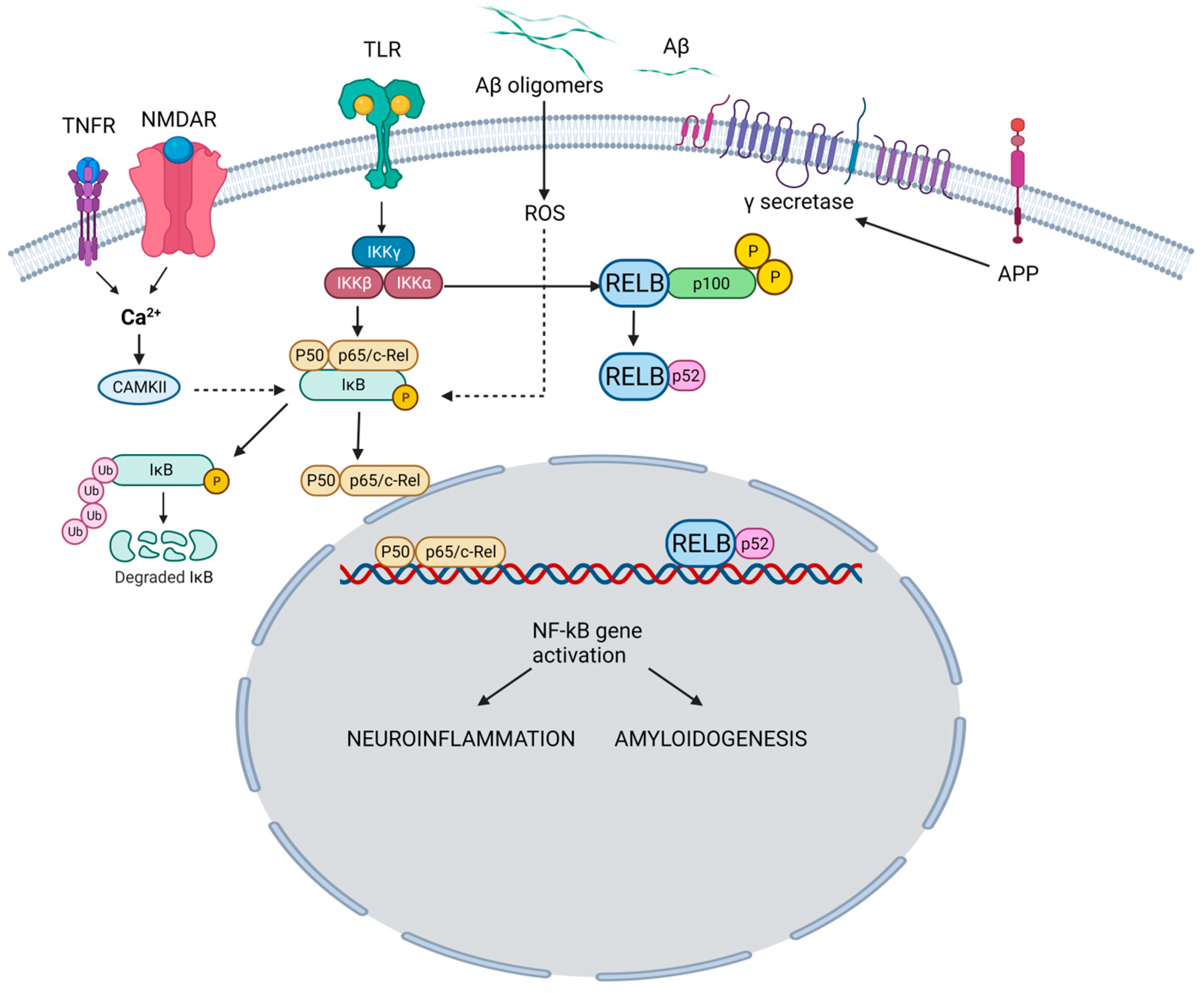
Figure 1. NF-κB pathway and its role in the pathogenesis of AD. In the canonical pathway, TNFR and NMDAR activation raises Ca2+, triggering CaMKII-mediated Ca2+-NF-κB linkage. Simultaneously, TLR activation acts on IKK subunits. Ca2+ leads to IκB kinase activation, IκB breakdown, and p65/p50 dimer formation. NF-κB enters the nucleus, binding neuronal gene targets (e.g., APP) for synaptic function, memory, and amyloid processing. Aβ, resulting from APP cleavage, activates ROS-dependent NF-κB, intensifying amyloid dysregulation. Pro-inflammatory agents (e.g., TNF-α, IL-1β) activate TLR through canonical (IKKβ-NEMO) and non-canonical (IKKγ, IKK-α) pathways. Both drive NF-κB-mediated neuroinflammation and neurodegeneration in AD. TNFR: Tumor necrosis factor receptor; NMDAR: N-Methyl-D-Aspartate Receptor; TLR: Toll-like receptor; Aβ: Amyloid beta; APP: Amyloid precursor protein; CAMKII: Calcium–calmodulin (CaM)-dependent protein kinase I; IKKγ: Inhibitor of nuclear factor-ĸB Kinase gamma (also known as NEMO: Nuclear factor-κB (NF-κB) essential modulator); IKKα: Inhibitor of nuclear factor-ĸB Kinase alpha; IKKβ: Inhibitor of nuclear factor-ĸB kinase beta; IĸB: Inhibitor of ĸB; ROS: Reactive oxygen species. TNF-α: Tumor necrosis factor-alpha, IL-1β: Interlukin-1 beta; RELB: Transcription factor.
Upon proteolytic processing, NF-κB1 generates the active p50 subunit. Similarly, NF-κB2 undergoes proteolytic processing to produce the active p52 subunit, which is involved in immune responses and lymphoid organ development. The p65 subunit aids in functional NF-κB dimer formation and is pivotal in regulating inflammation, immune responses, and cellular survival. The transcription factor, RelB, contributes to NF-κB dimer assembly and is particularly associated with immune responses, especially in lymphoid organ development. The c-Rel subunit modulates immune responses and gene expression related to inflammation and lymphoid organ development. These NF-κB family members form homo- or heterodimers, dictating target gene specificity and ensuing biological impacts [8].
Within neuroinflammation, NF-κB occupies a central role in coordinating the expression of genes that contribute to both protective and detrimental aspects of the inflammatory response in the nervous system. Primarily sequestered in an inactive state within the cytoplasm, NF-κB is bound to inhibitory proteins named IκBs. Upon activation, typically induced by a spectrum of stimuli encompassing pro-inflammatory cytokines, pathogens, or cellular stressors, these IκBs undergo phosphorylation and consequent degradation, facilitating the translocation of NF-κB to the nucleus. This nuclear migration empowers NF-κB to engage specific DNA sequences, thereby orchestrating the modulation of an extensive array of target genes. NF-κB signaling also influences the integrity of the blood–brain barrier (BBB), governing the passage of molecules between the bloodstream and the brain milieu. The activation of NF-κB in the endothelial cells of the BBB provokes heightened permeability, allowing immune cells and molecules to enter the brain [9].
Further, NF-κB activation in microglia triggers their transition from resting to activated states, releasing pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS). NF-κB is pivotal for initiating protective immune responses against pathogens and facilitating tissue repair. However, prolonged NF-κB activation within neurons can result in neurotoxicity, contributing to oxidative stress, excitotoxicity, and inflammation, all of which undermine neuronal well-being. NF-κB also influences the balance between neuronal survival and apoptosis. In some cases, NF-κB activation can promote the expression of anti-apoptotic genes, supporting neuronal survival [10]. However, chronic NF-κB activation can lead to a pro-apoptotic response in other situations.
2. Role of NF-κB Pathway Inhibitors in AD Treatment
The NF-κB pathway plays a significant role in neuroinflammation and other processes implicated in AD pathogenesis. Consequently, it has been considered a potential therapeutic target for the management of AD [11]. However, targeting the NF-κB pathway is complex due to its multifaceted role in promoting and regulating inflammation and immune responses. NF-κB pathway inhibitors offer the means to modulate this intricate signaling network and counteract neuroinflammation. By inhibiting NF-κB activation, these inhibitors can dampen the excessive release of pro-inflammatory molecules, thereby reducing the harmful impact of inflammation on neurons [12]. They hold the potential to alleviate neuronal damage and promote their survival in the face of AD-associated challenges.
Furthermore, NF-κB inhibitors may influence key pathological hallmarks of AD: Aβ plaques and tau tangles. NF-κB activation regulates enzymes involved in Aβ production and clearance [13]. Inhibiting this pathway could potentially modify Aβ metabolism, impacting the deposition and accumulation of Aβ plaques. Similarly, NF-κB activation has been linked to tau hyperphosphorylation and tangle formation. Inhibitors might intervene in these processes, attenuating tau pathology and promoting neuronal stability. However, it is essential to tread cautiously in inhibiting NF-κB, as this pathway also plays crucial roles in immune responses and cell survival [14]. A complete blockade could compromise the immune system’s ability to defend against pathogens and maintain tissue integrity. Striking the right balance between dampening neuroinflammation and preserving necessary immune functions is critical. The direct inhibition of NF-κB activation is a strategy under investigation for its therapeutic potential in various diseases, including AD [15]. Directly inhibiting NF-κB activation modulates the aberrant inflammatory response, potentially slowing disease progression. Researchers are exploring small molecules and compounds that directly target components of the NF-κB signaling pathway. These agents can interfere with the activation steps that lead to NF-κB translocation into the nucleus, where it regulates gene expression.
Advancements in NF-κB pathway inhibitor research are multifaceted. Scientists are striving to develop selective inhibitors that target specific pathway components, minimizing off-target effects. A study conducted by Elzayat and colleagues investigates mesenchymal stem cells (MSCs) and acitretin for AD treatment, focusing on the NF-kB pathway and miRNA regulation. Using a rat model, miR-146a, miR-155, necrotic, growth, and inflammatory genes were analyzed. Results showed that MSCs and acitretin restored normal levels, indicating potential therapeutic benefits. MiR-146a and miR-155 are proposed as AD biomarkers. This study underscores MSCs and acitretin’s capacity to modulate miRNAs and related genes in the NF-kB pathway, offering insights for future AD interventions [16].
Melatonin, a hormone the pineal gland produces, has been recognized for its role in regulating sleep–wake cycles and its potential anti-inflammatory properties. While melatonin’s direct impact on the NF-κB pathway is limited, it has been suggested that melatonin can indirectly modulate NF-κB activation through various mechanisms. Oxidative stress can activate NF-κB, leading to the expression of pro-inflammatory genes. Melatonin’s antioxidant properties indirectly inhibit NF-κB activation by reducing the triggering oxidative signals [17]. Melatonin can influence molecules downstream of the NF-κB pathway. For instance, it has been reported to downregulate COX-2 (Cyclooxygenase-2) expression, an inflammation enzyme that NF-κB often regulates. Melatonin’s neuroprotective effects may indirectly impact NF-κB activation. By preserving neuronal health and reducing neuroinflammation, melatonin can contribute to an environment where NF-κB activation is less pronounced. Merlo et al. demonstrated the counteracting effects of melatonin on the pro-inflammatory properties of lipopolysaccharides in in vitro models [18]. Another study investigated a phosphodiesterase 5 inhibitor (PDE5I) with dual antagonistic action on IKKB and TNFR1 to inhibit NF-kB and neuroinflammation. In silico docking with FDA-approved compounds identified avanafil with optimal IKKB and TNFR1 binding. Molecular dynamic studies confirmed avanafil’s stability with these targets. In a mouse model of lipopolysaccharide (LPS)-induced neuroinflammation and cognitive decline, avanafil at 6 mg/kg improved cognitive performance, reduced Aβ levels, and lowered inflammatory markers and oxidative parameters [19].
Bay 11-7082 is a synthetic small molecule that has gained attention for its potential as an NF-κB pathway inhibitor. This compound has shown inhibitory effects on IκB kinase (IKK), allowing NF-κB to translocate into the nucleus and initiate the expression of target genes involved in inflammation and immune responses. By inhibiting IKK and IκB degradation, Bay 11-7082 effectively interferes with interleukin-6 (IL-6) secretion and the subsequent activation of NF-κB, leading to decreased pro-inflammatory gene expression [20]. For Bay 11-7082 to effectively modulate NF-κB activity within the brain, it must cross the BBB. Developing strategies to enhance its delivery and distribution to the central nervous system while minimizing systemic effects is still a significant challenge [18]. Additionally, a low dose of recombinant IL-2 (aldesleukin) is currently under clinical trial for its clinical application in AD (NCT05821153; NCT05468073).
The SN50 analog is a cell-permeable, synthetic peptide derived from the NF-κB nuclear localization sequence, and it holds potential in AD treatment due to its ability to inhibit NF-κB nuclear translocation. A study conducted by Lin and colleagues suggests that the inhibitory effect is not due to disrupting the binding activity of NF-κB complexes but rather its ability to enter the cell and compete with NF-κB complexes for the cellular machinery responsible for nuclear translocation [21][22]. A study by Pogue and team reveals that upregulated miRNA-30b, associated with neuropathology in AD brain and LPS-stressed human neuronal–glial cells, targets neurofilament light (NF-L)-chain mRNA’s 3′-UTR. This results in the post-transcriptional downregulation of NF-L expression, observed in AD and LPS-treated cells, leading to neuronal cytoskeleton atrophy and synaptic disruption. MiRNA-30b is highly expressed in Aβ-treated models, facilitating LPS entry into neurons. Elevated miRNA-30b induces neuro-injury, inflammation, synaptic impairment, and neuron loss. This gut-microbiota-derived LPS-NF-kB-miRNA-30b-NF-L pathway links GI tract microbes’ LPS to AD’s neuropathology and synaptic disruption, offering insights into neurodegeneration mechanisms [23]. Optimizing dosages to achieve the desired therapeutic outcomes while avoiding potential side effects is an ongoing challenge. Table 1 enlists molecules being researched for their potential role in the NF-ĸB pathway in AD.
Table 1. The list of representative molecules targeting the NF-κB pathway to treat and manage Alzheimer’s disease.
| Molecule | NF-ĸB Target | Experimental Setting | Mechanism of Action | References |
|---|---|---|---|---|
| Alogliptin | Direct | In vitro | Modulates TLR4/MYD88/NF-κB and miRNA-155/SOCS-1 signaling pathways | [24] |
| AS62868 | Direct | In vitro | Inhibits IKKβ | [25] |
| Docosahexaenoic acid | Indirect | In vitro | Precursor to produce neuroprotection D1, a neuro-protecting agent in the CNS | [26] |
| Etanercept | Direct | RCT | TNF-α activity inhibitor | [27][28] |
| Forsythoside B | Direct | In vivo and in vitro | Reduces the serine 536 phosphorylation of IKKα/β, IκBα, and p65 | [29] |
| Minocycline | Direct | In vivo and in vitro | Reduction in IL-6; BACE inhibition | [30][31] |
| Pioglitazone | Indirect | In vitro | Downregulates glycogen synthase kinase 3 beta and cyclin-dependent kinase in microglia cells | [32][33] |
| PUFA-Plasmalogens | Direct | In vitro | Inhibits NF-kB, p38MAPK, and JNK pathways | [34] |
| Simufilam | Indirect | Inpatient | Reduces mTOR basal overactivity; restores the normal shape and function of Filamin A | [35] |
| Telmisartan | Indirect | In vivo and in vitro | AT1 blocking reduces IL-1β levels, resulting in anti-neuroinflammatory effects through JNK/c-Jun and NADPH oxidase pathways; partial PPAR-gamma-stimulating activity | [36][37][38] |
| TPCA-1 | Direct | In vivo | Inhibits IKKβ | [39] |
| VX-745 | Indirect | In vivo | p38 MAPKα inhibitor | [40] |
mTOR: Mammalian target of rapamycin; IKKβ: Nuclear factor kappa B kinase subunit beta; TNF-α: Tumor necrosis factor alpha; IL-6: Interleukin 6; BACE: β-Site amyloid precursor protein-cleaving enzyme; CNS: Central nervous system; NF-kB: Nuclear factor kappa B; p38MAPK: p38 mitogen-activated protein kinases; JNK: c-Jun N-terminal kinase; MYD88: Myeloid differentiation primary response protein 88; miRNA-155: MicroRNA-155; SOCS-1: Suppressor of cytokine signaling 1; IκBα: Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; p38 MAPKα: p38 mitogen-activated protein kinase alpha. RCT: Randomized clinical trials; AT1: Angiotensin 1; NADPH: Nicotinamide Adenine Dinucleotide Phosphate Hydrogen.
In practical terms, combining NF-κB inhibition with other therapeutic approaches may yield enhanced results. Research conducted by Shehata et al. aimed to formulate nanostructured lipid carriers (NLCs) that co-encapsulate donepezil and astaxanthin (DPL/AST–NLCs) and assess their effectiveness in AD-like rat models following daily intranasal administration [41]. AD is a complex disease with multiple underlying mechanisms, including Aβ aggregation, tau pathology, and synaptic dysfunction. Integrating NF-κB inhibitors with interventions targeting these aspects could offer a comprehensive treatment strategy, potentially addressing symptom relief and disease modification.
To translate NF-κB inhibition into clinical practice, robust clinical trials are imperative. These trials should involve diverse patient populations, incorporate appropriate outcome measures, and assess long-term safety. Furthermore, identifying reliable biomarkers that reflect NF-κB activity and its downstream effects will aid in monitoring treatment efficacy and patient response.
References
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569.
- Nelson, P.T.; Brayne, C.; Flanagan, M.E.; Abner, E.L.; Agrawal, S.; Attems, J.; Castellani, R.J.; Corrada, M.M.; Cykowski, M.D.; Di, J.; et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: Combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol. 2022, 144, 27–44.
- Uddin, M.S.; Kabir, M.T.; Jalouli, M.; Rahman, M.A.; Jeandet, P.; Behl, T.; Alexiou, A.; Albadrani, G.M.; Abdel-Daim, M.M.; Perveen, A.; et al. Neuroinflammatory Signaling in the Pathogenesis of Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 126–146.
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716.
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J. Exp. Med. 2017, 214, 1081–1092.
- Deolankar, S.C.; Patil, A.H.; Koyangana, S.G.; Subbannayya, Y.; Prasad, T.S.K.; Modi, P.K. Dissecting Alzheimer’s Disease Molecular Substrates by Proteomics and Discovery of Novel Post-translational Modifications. OMICS 2019, 23, 350–361.
- Fathi, N.; Mojtahedi, H.; Nasiri, M.; Abolhassani, H.; Yousefpour Marzbali, M.; Esmaeili, M.; Salami, F.; Biglari, F.; Rezaei, N. How do nuclear factor kappa B (NF-κB)1 and NF-κB2 defects lead to the incidence of clinical and immunological manifestations of inborn errors of immunity? Expert Rev. Clin. Immunol. 2023, 19, 329–339.
- Pan, W.; Deng, L.; Wang, H.; Wang, V.Y.F. Atypical IκB Bcl3 enhances the generation of the NF-κB p52 homodimer. Front. Cell Dev. Biol. 2022, 10, 930619.
- Yang, G.; Hu, Y.; Qin, X.; Sun, J.; Miao, Z.; Wang, L.; Ke, Z.; Zheng, Y. Micheliolide attenuates neuroinflammation to improve cognitive impairment of Alzheimer’s disease by inhibiting NF-κB and PI3K/Akt signaling pathways. Heliyon 2023, 9, e17848.
- Molina-Salinas, G.; Rodríguez-Chávez, V.; Langley, E.; Cerbon, M. Prolactin-induced neuroprotection against excitotoxicity is mediated via PI3K/AKT and GSK3β/NF-κB in primary cultures of hippocampal neurons. Peptides 2023, 166, 171037.
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095.
- Zhu, H.; Bai, Y.; Wang, G.; Su, Y.; Tao, Y.; Wang, L.; Yang, L.; Wu, H.; Huang, F.; Shi, H.; et al. Hyodeoxycholic acid inhibits lipopolysaccharide-induced microglia inflammatory responses through regulating TGR5/AKT/NF-κB signaling pathway. J. Psychopharmacol. 2022, 36, 849–859.
- Yang, S.; Magnutzki, A.; Alami, N.O.; Lattke, M.; Hein, T.M.; Scheller, J.S.; Kröger, C.; Oswald, F.; Yilmazer-Hanke, D.; Wirth, T.; et al. IKK2/NF-κB Activation in Astrocytes Reduces amyloid β Deposition: A Process Associated with Specific Microglia Polarization. Cells 2021, 10, 2669.
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780.
- Singh, S.; Singh, T.G. Role of Nuclear Factor Kappa B (NF-κB) Signalling in Neurodegenerative Diseases: An Mechanistic Approach. Curr. Neuropharmacol. 2020, 18, 918–935.
- Elzayat, E.M.; Shahien, S.A.; El-Sherif, A.A.; Hosney, M. Therapeutic potential of stem cells and acitretin on inflammatory signaling pathway-associated genes regulated by miRNAs 146a and 155 in AD-like rats. Sci. Rep. 2023, 13, 9613.
- Korkmaz, A.; Reiter, R.J.; Topal, T.; Manchester, L.C.; Oter, S.; Tan, D.X. Melatonin: An established antioxidant worthy of use in clinical trials. Mol. Med. 2009, 15, 43–50.
- Merlo, S.; Caruso, G.I.; Korde, D.S.; Khodorovska, A.; Humpel, C.; Sortino, M.A. Melatonin Activates Anti-Inflammatory Features in Microglia in a Multicellular Context: Evidence from Organotypic Brain Slices and HMC3 Cells. Biomolecules 2023, 13, 373.
- Chowdari Gurram, P.; Satarker, S.; Kumar, G.; Begum, F.; Mehta, C.; Nayak, U.; Mudgal, J.; Arora, D.; Nampoothiri, M. Avanafil mediated dual inhibition of IKKβ and TNFR1 in an experimental paradigm of Alzheimer’s disease: In silico and in vivo approach. J. Biomol. Struct. Dyn. 2022, 1–19.
- Cook, M.; Lin, H.; Mishra, S.K.; Wang, G.Y. BAY 11-7082 inhibits the secretion of interleukin-6 by senescent human microglia. Biochem. Biophys. Res. Commun. 2022, 617, 30–35.
- Choi, S.; Kim, J.H.; Roh, E.J.; Ko, M.J.; Jung, J.E.; Kim, H.J. Nuclear factor-kappaB activated by capacitative Ca2+ entry enhances muscarinic receptor-mediated soluble amyloid precursor protein (sAPPalpha) release in SH-SY5Y cells. J. Biol. Chem. 2006, 281, 12722–12728.
- Lin, Y.Z.; Yao, S.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of Nuclear Translocation of Transcription Factor NF-kappa B by a Synthetic Peptide Containing a Cell Membrane-permeable Motif and Nuclear Localization Sequence. J. Biol. Chem. 1995, 270, 14255–14258.
- Pogue, A.I.; Jaber, V.R.; Sharfman, N.M.; Zhao, Y.; Lukiw, W.J. Downregulation of Neurofilament Light Chain Expression in Human Neuronal-Glial Cell Co-Cultures by a Microbiome-Derived Lipopolysaccharide-Induced miRNA-30b-5p. Front. Neurol. 2022, 13, 900048.
- El-Sahar, A.E.; Shiha, N.A.; El Sayed, N.S.; Ahmed, L.A. Alogliptin Attenuates Lipopolysaccharide-Induced Neuroinflammation in Mice Through Modulation of TLR4/MYD88/NF-κB and miRNA-155/SOCS-1 Signaling Pathways. Int. J. Neuropsychopharmacol. 2020, 24, 158–169.
- Sarnico, I.; Boroni, F.; Benarese, M.; Alghisi, M.; Valerio, A.; Battistin, L.; Spano, P.; Pizzi, M. Targeting IKK2 by pharmacological inhibitor AS602868 prevents excitotoxic injury to neurons and oligodendrocytes. J. Neural Transm. 2008, 115, 693–701.
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.; Dua, P.; Lukiw, W.J. Regulation of Neurotropic Signaling by the Inducible, NF-kB-Sensitive miRNA-125b in Alzheimer’s Disease (AD) and in Primary Human Neuronal-Glial (HNG) Cells. Mol. Neurobiol. 2014, 50, 97–106.
- Clark, I. Basic Scientific Evidence Consistent with Etanercept Efficacy Against Alzheimer’s Disease. Neuroscience 2022, 484, 139.
- Butchart, J.; Brook, L.; Hopkins, V.; Teeling, J.; Püntener, U.; Culliford, D.; Sharples, R.; Sharif, S.; McFarlane, B.; Raybould, R.; et al. Etanercept in Alzheimer disease: A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology 2015, 84, 2161–2168.
- Kong, F.; Jiang, X.; Wang, R.; Zhai, S.; Zhang, Y.; Wang, D. Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-κB signaling in Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 305.
- Ferretti, M.T.; Allard, S.; Partridge, V.; Ducatenzeiler, A.; Cuello, A.C. Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer’s disease-like amyloid pathology. J. Neuroinflamm. 2012, 9, 62.
- Cheng, S.; Hou, J.; Zhang, C.; Xu, C.; Wang, L.; Zou, X.; Yu, H.; Shi, Y.; Yin, Z.; Chen, G. Minocycline reduces neuroinflammation but does not ameliorate neuron loss in a mouse model of neurodegeneration. Sci. Rep. 2015, 5, 10535.
- Alhowail, A.; Alsikhan, R.; Alsaud, M.; Aldubayan, M.; Rabbani, S.I. Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature. Drug Des. Devel. Ther. 2022, 16, 2919–2931.
- da Rocha, G.H.; Loiola, R.A.; de Paula-Silva, M.; Shimizu, F.; Kanda, T.; Vieira, A.; Gosselet, F.; Farsky, S.H.P. Pioglitazone Attenuates the Effects of Peripheral Inflammation in a Human In Vitro Blood-Brain Barrier Model. Int. J. Mol. Sci. 2022, 23, 12781.
- Youssef, M.; Ibrahim, A.; Akashi, K.; Hossain, M.S. PUFA-Plasmalogens Attenuate the LPS-Induced Nitric Oxide Production by Inhibiting the NF-kB, p38 MAPK and JNK Pathways in Microglial Cells. Neuroscience 2019, 397, 18–30.
- Wang, H.Y.; Pei, Z.; Lee, K.C.; Nikolov, B.; Doehner, T.; Puente, J.; Friedmann, N.; Burns, L.H. Simufilam suppresses overactive mTOR and restores its sensitivity to insulin in Alzheimer’s disease patient lymphocytes. Front. Aging 2023, 4, 1175601.
- Kurata, T.; Lukic, V.; Kozuki, M.; Wada, D.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Deguchi, K.; Yamashita, T.; Hishikawa, N.; et al. Long-term effect of telmisartan on Alzheimer’s amyloid genesis in SHR-SR after tMCAO. Transl. Stroke Res. 2015, 6, 107–115.
- Kishi, T.; Hirooka, Y.; Sunagawa, K. Telmisartan protects against cognitive decline via up-regulation of brain-derived neurotrophic factor/tropomyosin-related kinase B in hippocampus of hypertensive rats. J. Cardiol. 2012, 60, 489–494.
- Pang, T.; Wang, J.; Benicky, J.; Sánchez-Lemus, E.; Saavedra, J.M. Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-Jun and NADPH oxidase pathways. J. Neuroinflamm. 2012, 9, 102.
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969.
- Alam, J.J. Selective Brain-Targeted Antagonism of p38 MAPKα Reduces Hippocampal IL-1β Levels and Improves Morris Water Maze Performance in Aged Rats. J. Alzheimer’s Dis. 2015, 48, 219–227.
- Shehata, M.K.; Ismail, A.A.; Kamel, M.A. Combined Donepezil with Astaxanthin via Nanostructured Lipid Carriers Effective Delivery to Brain for Alzheimer’s Disease in Rat Model. Int. J. Nanomed. 2023, 18, 4193–4227.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
25 Sep 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No