Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Monica Embers | -- | 3034 | 2023-09-20 02:58:05 | | | |
| 2 | Catherine Yang | Meta information modification | 3034 | 2023-09-20 03:00:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Landry, R.L.; Embers, M.E. Pathogenesis of Multiple Sclerosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/49401 (accessed on 08 August 2026).
Landry RL, Embers ME. Pathogenesis of Multiple Sclerosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/49401. Accessed August 08, 2026.
Landry, Remi L., Monica E. Embers. "Pathogenesis of Multiple Sclerosis" Encyclopedia, https://encyclopedia.pub/entry/49401 (accessed August 08, 2026).
Landry, R.L., & Embers, M.E. (2023, September 20). Pathogenesis of Multiple Sclerosis. In Encyclopedia. https://encyclopedia.pub/entry/49401
Landry, Remi L. and Monica E. Embers. "Pathogenesis of Multiple Sclerosis." Encyclopedia. Web. 20 September, 2023.
Copy Citation
Multiple sclerosis (MS) is an immune inflammatory disease that causes demyelination of the white matter of the central nervous system. It is generally accepted that the etiology of MS is multifactorial and believed to be a complex interplay between genetic susceptibility, environmental factors, and infectious agents.
multiple sclerosis
Epstein–Barr virus (EBV)
human herpesvirus 6
varicella-zoster virus
1. Introduction
One of the key pathological features of MS is the presence of inflammatory lesions or plaques in the white matter of the CNS. These plaques are characterized by the infiltration of immune cells, such as T cells, B cells, and macrophages, into the CNS [1]. These cells release cytokines and other inflammatory mediators that activate resident glial cells, such as astrocytes and microglia, leading to a cascade of events that ultimately result in demyelination and axonal damage. The demyelination of axons in MS is thought to result from a combination of direct immune-mediated attack on myelin and the indirect effects of inflammation on oligodendrocytes, the cells responsible for producing myelin in the CNS. Demyelination leads to the exposure of axons, which can then become damaged due to a range of mechanisms, including oxidative stress, energy failure, and neurodegeneration that leads to brain atrophy [2]. The chronic inflammation associated with MS can also lead to the accumulation of gliosis in the CNS [3]. Gliosis is characterized by the proliferation of astrocytes and microglia, along with the deposition of extracellular matrix proteins such as collagen and fibronectin. The accumulation of tissue damage can disrupt the normal architecture of the CNS, and also impede remyelination and axonal regeneration. Plaques in the white matter are characterized by active destruction of axons and their myelin sheaths, which ultimately results in the development of irreversible neurological symptoms in the affected individual [1]. In addition to white matter lesions, MS can also affect the gray matter, particularly in later stages of disease. Gray matter atrophy is associated with cognitive impairment and disability in MS, and it is thought to result from a combination of axonal damage, neuronal loss, and gliosis [4].
2. Nature vs. Nurture
It is generally accepted that the etiology of MS is multifactorial and believed to be a complex interplay between genetic susceptibility, environmental factors such as smoking and obesity, and infectious agents that lead to immune dysregulation and CNS inflammation. It may be extremely valuable to define disease phenotypes based on the underlying pathologic mechanisms to achieve success with the personalized approach. Defining disease subtypes based on biology rather than on clinical manifestations should improve the validity of clinical trials, as drugs target the mechanism of disease and not the clinical stage. A recent study used machine learning to classify MS based on pathological features rather than disease subtype (RRMS, SPMS, and PPMS) for magnetic resonance imaging (MRI) [5]. The researchers used a training dataset of 6322 MS patients with every clinical subtype to define MRI-based subtypes and an independent cohort of 3068 patients for validation. Based on the earliest abnormalities, MS subtypes were defined as cortex-led, normal-appearing white matter-led, and lesion-led. People with the lesion-led subtype had the highest risk of confirmed disability progression and relapse rate [5]. People with the lesion-led MS subtype showed positive treatment responses in selected clinical trials [5]. These findings could provide important clues regarding etiology, either genetic or microbial.
While the exact cause of MS is not yet fully understood, it is believed that risk of developing the disease is strongly influenced by an individual’s genetic makeup. For decades, the only genes found to be risk factors were the human leukocyte antigen (HLA) genes. These genes strongly affect the immune reactivity of T cells. Certain variations of the HLA gene, such as HLA-DR2 and HLA-DQw1, have been linked to increased risk of developing MS, particularly in people of European descent [6][7]. HLA molecules play a pivotal role in presenting antigens to T cells. Certain haplotypes have been associated with increased susceptibility to disease, suggesting their involvement in initiating or perpetuating the autoimmune response against CNS components. Molecular mimicry adds a layer of intricacy to the HLA narrative [8]. Pathogens, which bear structural resemblances to self-antigens, can trigger cross-reactive immune responses. When considering HLA haplotypes, specific combinations might have a greater propensity to present both pathogen-derived and self-antigens, enhancing the likelihood of cross-reactivity [9][10]. This scenario could potentially fuel the characteristic autoimmune cascade of MS, as the immune system encounters both foreign and self-antigens through shared HLA context. The implications of this interplay are profound. Certain HLA haplotypes might act as conduits for molecular mimicry-driven cross-reactivity, inadvertently priming the immune system to target self-components in the CNS due to structural resemblances to pathogenic antigens [11]. This phenomenon could substantially influence disease susceptibility and progression, amplifying the autoimmune response against CNS tissues.
More recently, genome-wide association studies (GWAS) identified more than 200 genetic variants associated with increased MS risk, with approximately 30 associated with the major histocompatibility complex (MHC) locus [12]. Many of these variants are located in or near genes that are involved in immune function and regulation, such as the MHC region on chromosome 6, which contains several genes involved in antigen presentation and T-cell activation. Other genetic variants that have been identified via GWAS have well-ascribed functions in other aspects of immune regulation, such as cytokine signaling and T-cell differentiation, but some are associated with mitochondrial function or myelin structure. For example, variants in the interleukin 7 receptor (IL7R) gene have been shown to increase the risk of developing MS, likely by affecting T-cell homeostasis and activation [13][14][15]. Large-scale whole-genome sequencing studies have also identified rare genetic variants that are associated with increased risk of developing MS [16]. These variants are less common in the general population, but many are located in or near genes that are involved in immune function and regulation, similar to the common variants. Despite the progress made in identifying genetic variants associated with MS, the majority of the heritability of the disease remains unexplained. This suggests that there are likely many more genetic variants that contribute to MS risk, and that the genetic architecture of the disease is likely to be very complex.
In addition to identifying variants in immune function that contribute to MS risk, genetic studies have also provided insight into the biology of the disease. These genetic factors are not sufficient on their own to cause MS, and environmental factors such as viral infections, smoking, and vitamin D deficiency also play important roles in disease development (Figure 1). Factors that exhibit correlations with certain ethnic groups or MS subtypes have emerged as intriguing associations that reflect the intricate interplay between genetic predisposition, environmental influences, and disease susceptibility. Variants in genes involved in vitamin D metabolism and regulation have been associated with increased risk of developing MS, suggesting that vitamin D deficiency may play a role in the disease. Studies have shown that low levels of vitamin D are associated with increased risk of developing MS and that vitamin D supplementation may help to reduce disease activity and disability in people with MS [17]. Noteworthy observations point to a higher prevalence of MS among individuals of Northern European descent, attributed in part to paler skin facilitating efficient vitamin D synthesis upon sunlight exposure [17]. Smoking is another environmental factor that has been linked to increased risk of developing MS. Studies have shown that people who smoke have a higher risk of developing MS, and that smoking may also contribute to more severe disease progression [18]. Diet has also been proposed as a potential environmental factor in MS etiology. The intriguing prospect of the gut microbiota impacting disease subtype underscores its potential to modulate MS progression differentially across patient groups. Cultural dietary practices often exhibit ethnic nuances, which extend to their influence on the gut microbiota composition. A diet high in saturated fat and low in fruits and vegetables has been associated with increased risk of developing MS, while a diet rich in omega-3 fatty acids and antioxidants may help to reduce disease activity and disability [19]. As research advances, a refined understanding of these complex relationships holds the potential to unveil novel insights into the mechanisms underlying MS etiology, ethnic disparities, and disease subtypes.
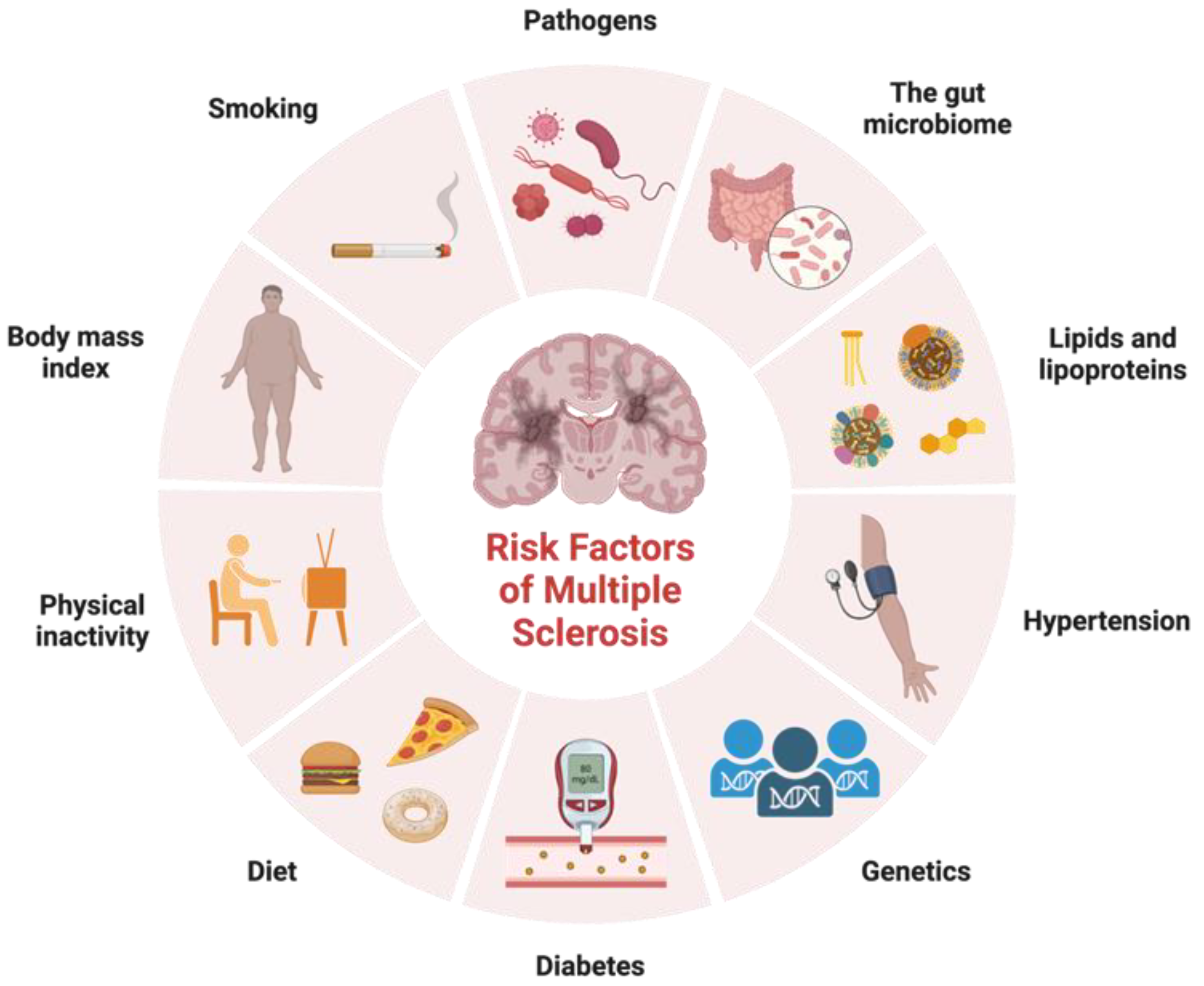
Figure 1. Risk factors for multiple sclerosis. Adapted from “Risk Factors of Dementia”, by BioRender.com. Accessed on 17 February 2023. Retrieved from https://app.biorender.com/biorender-templates.
3. Microbes and Infectious Agents
Among the environmental factors that have been implicated in the development of MS are infectious agents and microbes. The gut microbiome, which refers to the community of microorganisms that inhabit the human gastrointestinal tract, has been increasingly recognized as an important player in various physiological and pathological processes, including immune regulation, metabolism, and neurological function. The gut microbiome is known to interact with the host immune system through gut-associated lymphoid tissue (GALT), which is the largest immune organ in the body and implicated in the pathogenesis of various autoimmune and inflammatory diseases, including MS [20]. Recent studies have shown that the gut microbiome in MS patients differs from that in healthy individuals, with alterations in the abundance and diversity of certain bacterial species. For instance, MS patients have been shown to have decreased abundances of some beneficial bacteria, such as Akkermansia muciniphila, Faecalibacterium prausnitzii, and Butyricimonas spp., and increased abundances of some pro-inflammatory bacteria, such as Eubacterium hallii, Ruminococcus gnavus, and Parabacteroides distasonis [21][22]. These alterations in the gut microbiome have been proposed to contribute to the development and progression of MS through several mechanisms, including modulation of the immune response, alteration of blood–brain barrier (BBB) integrity, and production of neuroactive metabolites [23]. The gut microbiota and its prominent metabolic products, known as short-chain fatty acids (SCFAs), stand as pivotal entities in maintaining gut homeostasis and have implications in metabolic disease occurrence. SCFAs, such as acetate, butyrate, and propionate, are formed by gut microbial fermentation of dietary fibers and carbohydrates [24]. These SCFAs play an immunomodulatory role, with potential to affect the CNS. These substances can play roles in regulating blood pressure, GI function, and immune system function [25] Interestingly, decreased SCFA levels have been reported in patients with diseases such as Parkinson’s, Alzheimer’s, and anorexia nervosa [26]. In individuals afflicted by MS, there also appears to be an impact on the enteric nervous system (ENS). The ENS occupies a central role in orchestrating the intricate interplay between the gut microbiota and gastrointestinal homeostasis. The ENS plays an essential role in maintaining gut barrier integrity and immune surveillance [27]. SCFAs can influence ENS function. Notably, the ENS, constituting a substantial portion of the peripheral nervous system, remarkably mirrors the components and functionalities of the CNS. Signs of ENS disorder have been found in various neurodegenerative conditions, such as amyotrophic lateral sclerosis (ALS), Alzheimer’s, Parkinson’s, and MS [28]. Given the potential role of the gut microbiome in MS, there has been growing interest in developing microbiome-based therapies for this disease. These approaches include the use of probiotics, prebiotics, fecal microbiota transplantation, and dietary interventions aimed at modulating the gut microbiome. However, further research is needed to better understand the complex interactions between the gut microbiome and MS and to identify the most effective and safe microbiome-based interventions for this disease. Mucosal-associated invariant T (MAIT) cells also play a role in the gut–brain axis and the development of autoimmune neuropathology [29]. A recent study demonstrated that MAIT cells were significantly more activated in people with MS compared to healthy donors in response to yeast strains isolated from fecal samples. In addition, immunofluorescent staining of post-mortem brain tissues from individuals with the secondary progressive form of MS showed that MAIT cells cross the BBB and produce pro-inflammatory cytokines in the brain [30]. The process appears to begin with dysbiosis in the gut, followed by microbe-induced activation of MAIT cells in a TCR-dependent or independent manner. These cells become activated by IL-23 released from dendritic cells and monocytes. The activated MAIT cells then migrate to the CNS, cross the BBB, and release pro-inflammatory cytokines such as IL-17, GM-CSF, and IFN-ɣ, leading to neuronal damage [31]. MAIT cells also secrete CCL20, which recruits other CCR6-expressing T cells across the BBB, exacerbating neuroinflammation [32].
In addition to the gut microbiome, several studies have implicated microbial infections as determinants of MS risk. An ongoing debate persists as to whether microbial infections trigger MS in genetically predisposed individuals. Epigenetic mechanisms in the context of MS constitute a captivating terrain of research, and their intricate interplay with microbial involvement offers a multifaceted perspective on the disease’s etiopathogenesis [33]. Epigenetics involves modifications to DNA and histone proteins that orchestrate gene expression patterns without altering the genetic sequence. Epigenetic mechanisms assume a pivotal role in mediating the interplay between genetic susceptibility and environmental influences. Microbial involvement has been shown to modulate epigenetic markers [34]. DNA methylation, a prominent epigenetic modification, entails the addition of methyl groups to DNA strands, often resulting in gene silencing [35]. In the context of microbial exposure, DNA methylation patterns can be dynamically altered, impacting genes associated with immune responses and inflammation [36]. Microbial interactions might provoke epigenetic changes that skew the immune balance, potentially contributing to the immune dysregulation that characterizes MS. Histone modifications offer another layer of epigenetic orchestration. Microbial involvement has been shown to modulate epigenetic markers [34]. This modulation might significantly impact the equilibrium between regulatory and pro-inflammatory responses relevant to MS pathogenesis. Non-coding RNAs, encompassing microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), stand as another dimension of epigenetic modulation influenced by microbes [37]. These molecules, although they do not encode proteins themselves, profoundly impact gene expression by binding to messenger RNAs, either inhibiting or facilitating their translation. Perturbed miRNAs and lncRNAs, sculpted in response to microbial cues, might markedly contribute to the immune dysregulation that characterizes MS.
The role of infectious agents is also supported by the “hygiene hypothesis,” which proposes that reduced exposure to pathogens early in life can increase the risk for autoimmune diseases like MS [38]. This is due to an imbalance in Th1 and Th2 immune responses. However, helminths and protozoans can act with different immunological mechanisms. Protozoans induce a strong Th1 response through the production of pro-inflammatory mediators such as IL-12, nitric oxide, and IFN-γ [39]. Helminths, on the other hand, induce a Th2 response characterized by the release of IL-4, IL-5, and IL-10 [40]. The infectious origin of MS considers whether infections induce or accelerate autoimmune diseases like MS. Numerous infectious agents have been suggested to play roles in the pathogenesis of MS, including viruses, bacteria, and parasites. Among the most widely studied are Epstein–Barr virus (EBV), human herpesvirus 6 (HHV-6), varicella-zoster virus (VZV), Chlamydia pneumoniae, and Helicobacter pylori; however, to date, no infectious agents have been proven to cause MS.
4. Treatments
Over the years, various treatments have been developed to manage MS, aiming to reduce relapses, slow disease progression, and alleviate symptoms. These treatments can be broadly categorized into disease-modifying therapies (DMTs) and symptomatic treatments [41][42]. Symptomatic treatments for MS aim to alleviate and manage specific symptoms, such as spasticity, pain, bladder dysfunction, and fatigue. These treatments include muscle relaxants, beta blockers, physical therapy, pain relievers, medications to improve bladder control, speech therapy, and anti-fatigue medications.
DMTs have revolutionized MS management by targeting aberrant immune responses. Their mechanisms of action vary, encompassing immunomodulation, immunosuppression, and immune reconstitution. Monoclonal antibodies (MABs) have gained significant attention for their precision in targeting immune cells and molecules involved in MS pathogenesis [43]. Interferon beta was one of the earliest DMTs approved for MS. Interferon beta is available in different formulations, including interferon beta-1a and interferon beta-1b, and works by modulating the immune response, reducing inflammation, and promoting a more balanced immune system [44]. It also helps to decrease the number and severity of relapses in RRMS. Ocrelizumab is another monoclonal antibody that targets B cells, a type of immune cell involved in the immune response that damages myelin. By depleting certain B cells, ocrelizumab has demonstrated efficacy in reducing relapse rates and delaying progression in RRMS and PPMS [43]. Other DMTs, like natalizumab and alemtuzumab, have shown promising results by modulating immune cell migration and function. Natalizumab was first approved in 2004 and is a monoclonal antibody that targets integrins on immune cells, specifically preventing immune cells from crossing the BBB and entering the CNS, and therefore reducing the inflammation seen in MS [45]. Alemtuzumab targets CD52, a protein found on the surface of various immune cells [43]. It leads to the depletion of lymphocytes, particularly T and B cells, followed by repopulation with new cells. This “resetting” of the immune system can help to reduce inflammation and the autoimmune response seen in MS. Glatiramer acetate, commonly known as Copaxone, is another DMT that is thought to work by altering the immune response [46]. It is believed to simulate myelin basic protein, a component of the myelin sheath that is targeted in MS. By doing so, it diverts the immune system’s attack away from myelin, helping to reduce inflammation and the frequency of relapses. Dimethyl fumarate is an oral DMT that exerts its effects through multiple mechanisms, including activation of the Nrf2 pathway, which is involved in cellular defense against oxidative stress [47][48]. This drug helps to reduce inflammation and oxidative damage in the CNS. Fingolimod was the first DMT approved for relapsing forms of MS [49]. It is an oral DMT that works by binding to sphingosine-1-phosphate receptors on immune cells, preventing them from leaving lymph nodes and entering the central nervous system [49]. This reduces the number of immune cells available to cause inflammation in the brain and spinal cord.
The aforementioned DMTs can have implications for how the immune system responds to infectious insult. Ocrelizumab, alemtuzumab, and fingolimod can suppress the immune system to varying degrees. However, the immunomodulatory effects of DMTs can inadvertently impact the host’s ability to respond to microorganisms, influence the reactivation of latent infections, and potentially increase susceptibility to various types of viral, bacterial, and fungal infections. Some DMTs, like natalizumab, can affect the immune system’s response to specific viral infections, particularly the John Cunningham virus (JC virus) [50]. This virus can lead to progressive multifocal leukoencephalopathy (PML), a rare but serious brain infection [50]. Other immunosuppressive DMTs may disturb immune surveillance, triggering reactivation of latent infections. Varicella-zoster virus and tuberculosis are notable examples of infections that can reactivate due to compromised immune control [51]. The complex interplay between MS DMTs, microorganisms, and latent infections underscores the need for a comprehensive understanding of both therapeutic benefits and potential immunological risks.
References
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum 2013, 19, 901–921.
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37.
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687.
- Messina, S.; Patti, F. Gray matters in multiple sclerosis: Cognitive impairment and structural MRI. Mult. Scler. Int. 2014, 2014, 609694.
- Eshaghi, A.; Young, A.L.; Wijeratne, P.A.; Prados, F.; Arnold, D.L.; Narayanan, S.; Guttmann, C.R.G.; Barkhof, F.; Alexander, D.C.; Thompson, A.J.; et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat. Commun. 2021, 12, 2078.
- Cook, S.D. Multiple sclerosis and viruses. Mult. Scler. 1997, 3, 388–389.
- Jersild, C.; Dupont, B.; Fog, T.; Platz, P.J.; Svejgaard, A. Histocompatibility determinants in multiple sclerosis. Transplant. Rev. 1975, 22, 148–163.
- Libbey, J.E.; McCoy, L.L.; Fujinami, R.S. Molecular mimicry in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 127–147.
- Liblau, R.; Gautam, A.M. HLA, molecular mimicry and multiple sclerosis. Rev. Immunogenet. 2000, 2, 95–104.
- Ruhl, G.; Niedl, A.G.; Patronov, A.; Siewert, K.; Pinkert, S.; Kalemanov, M.; Friese, M.A.; Attfield, K.E.; Antes, I.; Hohlfeld, R.; et al. Multiple sclerosis: Molecular mimicry of an antimyelin HLA class I restricted T-cell receptor. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e241.
- Rojas, M.; Restrepo-Jimenez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123.
- Cotsapas, C.; Mitrovic, M. Genome-wide association studies of multiple sclerosis. Clin. Transl. Immunol. 2018, 7, e1018.
- Galarza-Munoz, G.; Briggs, F.B.S.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human Epistatic Interaction Controls IL7R Splicing and Increases Multiple Sclerosis Risk. Cell 2017, 169, 72–84.e13.
- Hoe, E.; McKay, F.C.; Schibeci, S.D.; Gandhi, K.; Heard, R.N.; Stewart, G.J.; Booth, D.R. Functionally significant differences in expression of disease-associated IL-7 receptor alpha haplotypes in CD4 T cells and dendritic cells. J. Immunol. 2010, 184, 2512–2517.
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007, 39, 1083–1091.
- Patsopoulos, N.A. Genetics of Multiple Sclerosis: An Overview and New Directions. Cold Spring Harb. Perspect. Med. 2018, 8, a028951.
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol. Ther. 2018, 7, 59–85.
- Nishanth, K.; Tariq, E.; Nzvere, F.P.; Miqdad, M.; Cancarevic, I. Role of Smoking in the Pathogenesis of Multiple Sclerosis: A Review Article. Cureus 2020, 12, e9564.
- Stoiloudis, P.; Kesidou, E.; Bakirtzis, C.; Sintila, S.A.; Konstantinidou, N.; Boziki, M.; Grigoriadis, N. The Role of Diet and Interventions on Multiple Sclerosis: A Review. Nutrients 2022, 14, 1150.
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141.
- Kadowaki, A.; Quintana, F.J. The Gut-CNS Axis in Multiple Sclerosis. Trends Neurosci. 2020, 43, 622–634.
- Boussamet, L.; Rajoka, M.S.R.; Berthelot, L. Microbiota, IgA and Multiple Sclerosis. Microorganisms 2022, 10, 617.
- Tang, W.; Zhu, H.; Feng, Y.; Guo, R.; Wan, D. The Impact of Gut Microbiota Disorders on the Blood-Brain Barrier. Infect. Drug Resist. 2020, 13, 3351–3363.
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105.
- Wu, Y.; Xu, H.; Tu, X.; Gao, Z. The Role of Short-Chain Fatty Acids of Gut Microbiota Origin in Hypertension. Front. Microbiol. 2021, 12, 730809.
- Vijay, A.; Valdes, A.M. Role of the gut microbiome in chronic diseases: A narrative review. Eur. J. Clin. Nutr. 2022, 76, 489–501.
- Luo, S.; Zhu, H.; Zhang, J.; Wan, D. The Pivotal Role of Microbiota in Modulating the Neuronal-Glial-Epithelial Unit. Infect. Drug Resist. 2021, 14, 5613–5628.
- Rao, M.; Gershon, M.D. The bowel and beyond: The enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528.
- Mechelli, R.; Romano, S.; Romano, C.; Morena, E.; Buscarinu, M.C.; Bigi, R.; Bellucci, G.; Renie, R.; Pellicciari, G.; Salvetti, M.; et al. MAIT Cells and Microbiota in Multiple Sclerosis and Other Autoimmune Diseases. Microorganisms 2021, 9, 1132.
- Gargano, F.; Guerrera, G.; Piras, E.; Serafini, B.; Di Paola, M.; Rizzetto, L.; Buscarinu, M.C.; Annibali, V.; Vuotto, C.; De Bardi, M.; et al. Proinflammatory mucosal-associated invariant CD8+ T cells react to gut flora yeasts and infiltrate multiple sclerosis brain. Front. Immunol. 2022, 13, 890298.
- Schnell, A.; Huang, L.; Singer, M.; Singaraju, A.; Barilla, R.M.; Regan, B.M.L.; Bollhagen, A.; Thakore, P.I.; Dionne, D.; Delorey, T.M.; et al. Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 2021, 184, 6281–6298.e23.
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Martin-Orozco, N.; Kang, H.S.; Ma, L.; Panopoulos, A.D.; et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 2008, 181, 8391–8401.
- Gacias, M.; Casaccia, P. Epigenetic Mechanisms in Multiple Sclerosis. Rev. Esp. Escler. Mult. 2014, 6, 25–35.
- Woo, V.; Alenghat, T. Epigenetic regulation by gut microbiota. Gut Microbes 2022, 14, 2022407.
- Lanata, C.M.; Chung, S.A.; Criswell, L.A. DNA methylation 101: What is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci. Med. 2018, 5, e000285.
- Qin, W.; Scicluna, B.P.; van der Poll, T. The Role of Host Cell DNA Methylation in the Immune Response to Bacterial Infection. Front. Immunol. 2021, 12, 696280.
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target Oncol. 2020, 15, 261–278.
- Correale, J.; Farez, M.F.; Gaitan, M.I. Environmental factors influencing multiple sclerosis in Latin America. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317715049.
- Gurung, P.; Kanneganti, T.D. Immune responses against protozoan parasites: A focus on the emerging role of Nod-like receptors. Cell. Mol. Life Sci. 2016, 73, 3035–3051.
- Nutman, T.B. Looking beyond the induction of Th2 responses to explain immunomodulation by helminths. Parasite Immunol. 2015, 37, 304–313.
- Higuera, L.; Carlin, C.S.; Anderson, S. Adherence to Disease-Modifying Therapies for Multiple Sclerosis. J. Manag. Care Spec. Pharm. 2016, 22, 1394–1401.
- Robertson, D.; Moreo, N. Disease-Modifying Therapies in Multiple Sclerosis: Overview and Treatment Considerations. Fed. Pract. 2016, 33, 28–34.
- Voge, N.V.; Alvarez, E. Monoclonal Antibodies in Multiple Sclerosis: Present and Future. Biomedicines 2019, 7, 20.
- Filipi, M.; Jack, S. Interferons in the Treatment of Multiple Sclerosis: A Clinical Efficacy, Safety, and Tolerability Update. Int. J. MS Care 2020, 22, 165–172.
- Yaldizli, O.; Putzki, N. Natalizumab in the treatment of multiple sclerosis. Ther. Adv. Neurol. Disord. 2009, 2, 115–128.
- Jalilian, B.; Einarsson, H.B.; Vorup-Jensen, T. Glatiramer acetate in treatment of multiple sclerosis: A toolbox of random co-polymers for targeting inflammatory mechanisms of both the innate and adaptive immune system? Int. J. Mol. Sci. 2012, 13, 14579–14605.
- Prosperini, L.; Haggiag, S.; Ruggieri, S.; Tortorella, C.; Gasperini, C. Dimethyl Fumarate or Teriflunomide for Relapsing-Remitting Multiple Sclerosis: A Meta-analysis of Post-marketing Studies. Neurotherapeutics 2023, 20, 1275–1283.
- Prosperini, L.; Pontecorvo, S. Dimethyl fumarate in the management of multiple sclerosis: Appropriate patient selection and special considerations. Ther. Clin. Risk Manag. 2016, 12, 339–350.
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18.
- Sgarlata, E.; Chisari, C.G.; Toscano, S.; Finocchiaro, C.; Lo Fermo, S.; Millefiorini, E.; Patti, F. Changes in John Cunningham Virus Index in Multiple Sclerosis Patients Treated with Different Disease-Modifying Therapies. Curr. Neuropharmacol. 2022, 20, 1978–1987.
- Sedighi, S.; Gholizadeh, O.; Yasamineh, S.; Akbarzadeh, S.; Amini, P.; Favakehi, P.; Afkhami, H.; Firouzi-Amandi, A.; Pahlevan, D.; Eslami, M.; et al. Comprehensive Investigations Relationship between Viral Infections and Multiple Sclerosis Pathogenesis. Curr. Microbiol. 2022, 80, 15.
More
Information
Subjects:
Orthopedics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
20 Sep 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No