Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Leonid A. Shaposhnikov | -- | 4830 | 2023-09-19 22:13:59 | | | |
| 2 | Peter Tang | + 1 word(s) | 4831 | 2023-09-20 04:37:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shaposhnikov, L.A.; Savin, S.S.; Tishkov, V.I.; Pometun, A.A. Ribonucleoside Hydrolase Structure. Encyclopedia. Available online: https://encyclopedia.pub/entry/49396 (accessed on 26 July 2026).
Shaposhnikov LA, Savin SS, Tishkov VI, Pometun AA. Ribonucleoside Hydrolase Structure. Encyclopedia. Available at: https://encyclopedia.pub/entry/49396. Accessed July 26, 2026.
Shaposhnikov, Leonid A., Svyatoslav S. Savin, Vladimir I. Tishkov, Anastasia A. Pometun. "Ribonucleoside Hydrolase Structure" Encyclopedia, https://encyclopedia.pub/entry/49396 (accessed July 26, 2026).
Shaposhnikov, L.A., Savin, S.S., Tishkov, V.I., & Pometun, A.A. (2023, September 19). Ribonucleoside Hydrolase Structure. In Encyclopedia. https://encyclopedia.pub/entry/49396
Shaposhnikov, Leonid A., et al. "Ribonucleoside Hydrolase Structure." Encyclopedia. Web. 19 September, 2023.
Copy Citation
Ribonucleoside hydrolases are enzymes that catalyze the cleavage of ribonucleosides to nitrogenous bases and ribose. These enzymes are found in many organisms: bacteria, archaea, protozoa, metazoans, yeasts, fungi and plants.
nucleoside hydrolases
crystal structure
catalysis mechanism
enzyme kinetics
cancer drug design
1. Introduction
With technological progress, the sequencing of the genomes of various organisms has turned from an expensive and rare procedure into an affordable and routine one. Currently, many genomes of various organisms from all three kingdoms have been sequenced, in which key proteins and enzymes with important functions have been annotated. However, the study of the key enzymes of such a vast set of organisms is a laborious process, because it is important not only to find the enzyme but also to study its main properties in order to understand and describe its physiological role and propose a potentially useful application for humans, be it in biotechnology, diagnostics, or medicine. In addition, some enzymes go unnoticed for a long time, because their potential benefits were not always clear at first glance and the studies carried out were not systematized.
2. Physiological Role of Rih Family Proteins
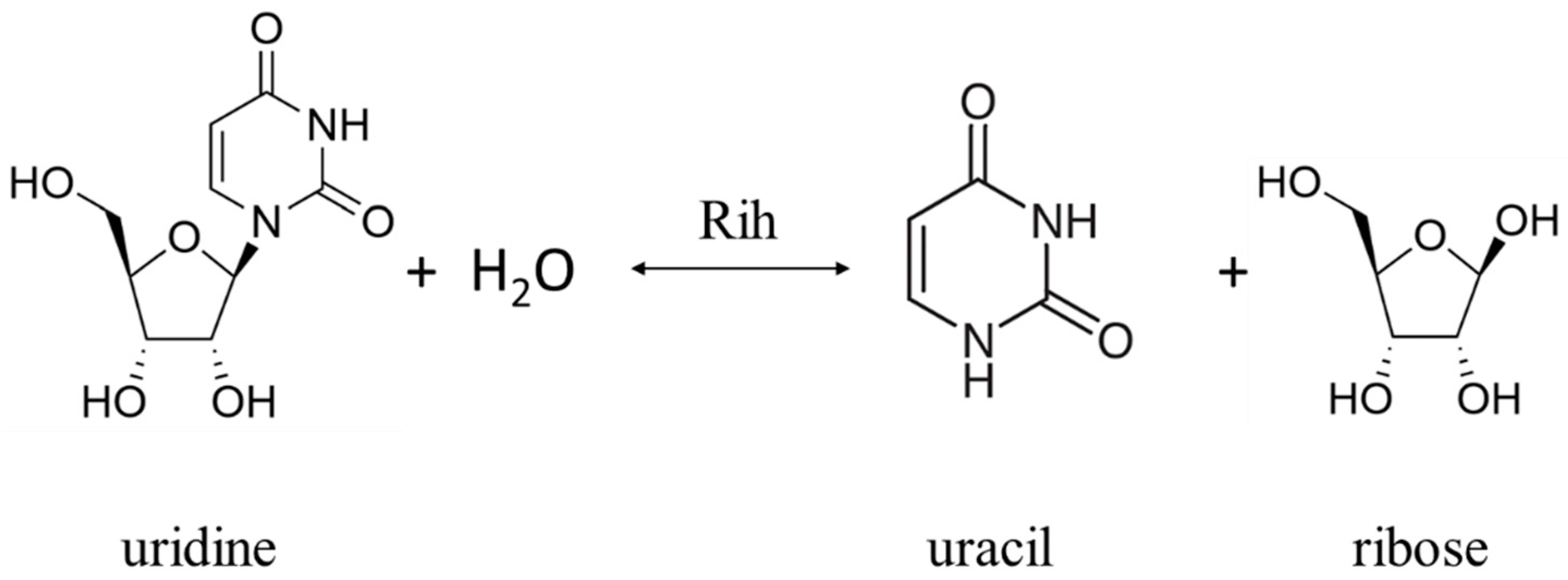
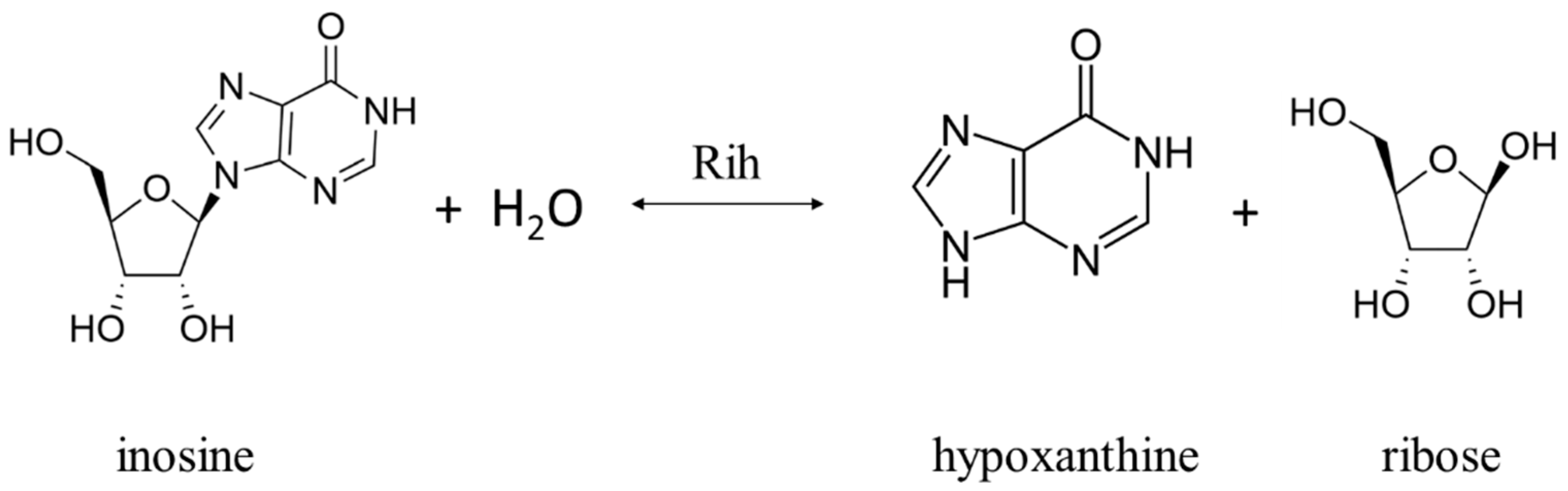
Ribonucleoside hydrolases (Rih) (EC 3.2.2.1–3.2.2.3, 3.2.2.7, 3.2.2.8, 3.2.2.13, 3.2.2.25) are a family of enzymes that are able to hydrolyze ribonucleosides at the N glycosidic bond to ribose and the corresponding nitrogenous base (Figure 1 and Figure 2). All chemical structures in this work from here onwards were drawn using the free online service ChemDraw JS [1].
Figure 1. Uridine cleavage reaction by pyrimidine-specific and nonspecific Rih hydrolases.
Figure 2. Inosine cleavage reaction by purine-specific and nonspecific Rih hydrolases.
For protozoan parasites (e.g., Trypanosoma brucei brucei, Leishmania major, Crithidia fasciculata), the role of these enzymes in metabolism is clear. These organisms cannot synthesize nitrogenous bases de novo; they must obtain them in other ways. To do this, they receive ribonucleosides from the outside, which are further converted into nitrogenous bases by phosphorylation or by hydrolysis. To implement the second process, their genome contains genes encoding certain Rih hydrolases, and thus for these organisms, such hydrolases are a key participant in metabolism [2][3][4][5][6][7][8][9][10][11][12][13][14][15][16].
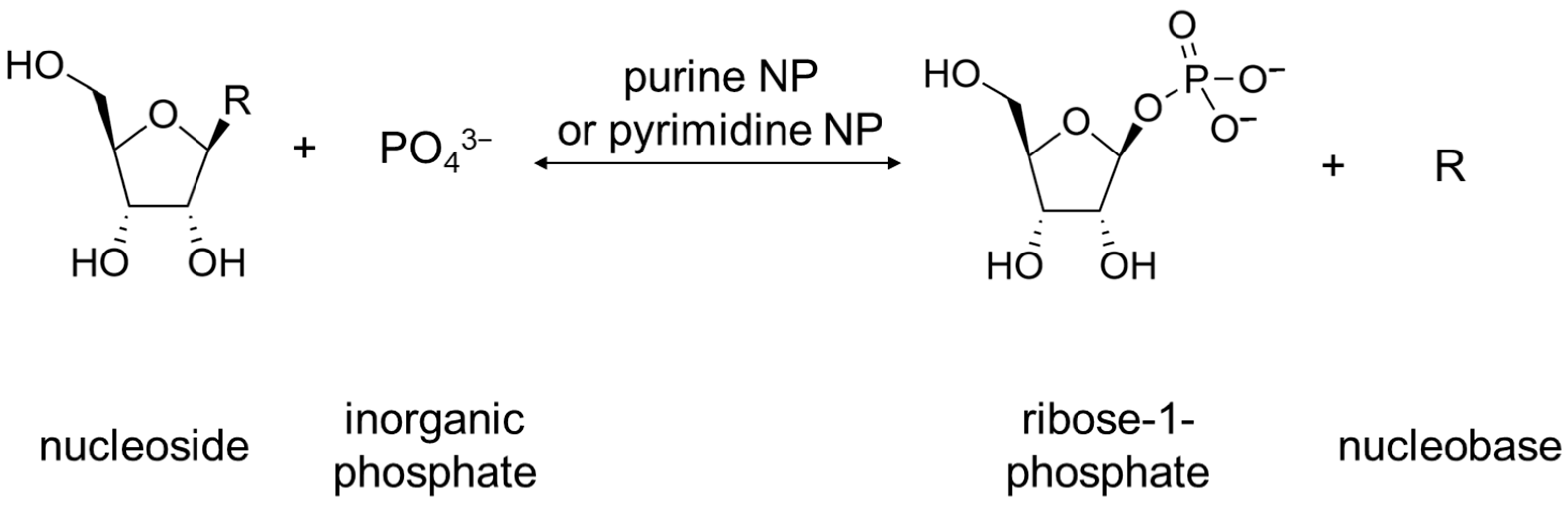
For bacteria, the physiological role of these enzymes is not completely clear. Bacteria have two common metabolic pathways for the production of purines and pyrimidines: de novo synthesis and recycling of nitrogenous bases and nucleosides. It is noteworthy that neither de novo synthesis nor the recycling route involves a simple synthesis of nucleosides from nitrogenous bases and ribose. Instead, the synthesis of nucleosides from scratch occurs in several steps, and the purine or pyrimidine bases are synthesized from several precursor molecules [17] (pp. 2883–2897). During the reutilization (salvage) path, phosphorylation and phosphorylases associated with this process play an important role [17] (p. 2916). Purine and pyrimidine nucleoside phosphorylases (purine NP, EC 2.4.2.1; pyrimidine NP EC 2.4.2.2) play a key role in the salvage pathway of nitrogenous bases [18][19]. These enzymes catalyze the reaction shown below in Figure 3.
Figure 3. Reaction catalyzed by purine NP and pyrimidine NP. R can be either purine nucleobase or pyrimidine nucleobase.
Curiously enough, these enzymes are more commonly used in different organisms as opposed to nucleoside hydrolases for purine and pyrimidine salvage pathways, the exception being protozoa, where nucleoside hydrolases play a major role in metabolism. While the reactions of these different types of enzymes (NHs and NPs) are different, the common role is the same: making a nitrogenous base from a nucleoside. Still, only NPs are more commonly used in organisms for salvage of purines and pyrimidines, although genes of NHs are present.
According to this, ribonucleoside hydrolases do not play a key role in the metabolism of bacteria, although these genes are present in their genomes, though some spore-forming bacteria, such as Bacillus cereus and Bacillus anthracis, require nucleoside hydrolases to prevent adenosine- or inosine-induced sporulation by cleaving the appropriate nucleosides [20][21][22].
For plants, nucleoside hydrolases may be needed under stressful conditions with a lack of nitrogen, when nitrogenous bases become its source, for the production of which Rih hydrolases is needed. Under normal conditions, these enzymes do not play a key role. These hydrolases have been found in various plants [23][24][25][26][27], and in some plants these enzymes are present as several isoforms. The physiological role of Rih hydrolases in plants is not limited to the cleavage of standard ribonucleosides. Decreased nucleoside hydrolase activity in Physcomitrella patens leads to delayed bud formation, suggesting involvement of Rih hydrolases in cytokinin metabolism [23].
In the case of other eukaryotes, nucleoside hydrolases have been found in yeast [28], fungi [29][30][31][32][33], nematodes [34][35], insects [36], and fish [37]. In addition, genes homologous to various Rih hydrolases have been found in other eukaryotes, but the enzymes encoded by these genes have not been studied. In mammals, Rih genes have not yet been found. Nucleoside hydrolase from the Aedes aegypti mosquito, which is a carrier of the Dengue fever virus, is injected into the body at the site of a mosquito bite, destroying circulating adenosine, thereby preventing activation of mast cells through purinergic signaling and thus acting as a local anesthetic [36]. The yeast Saccharomyces cerevisiae requires nucleoside hydrolase in the metabolism of pyridine nucleoside nicotinamide riboside, a precursor of NAD+ [38]. Nucleoside hydrolase activity in this case contributes to the activation of sirtuins, which are important for prolonging the life of yeast cells. In other eukaryotic organisms, the role of Rih hydrolases remains unclear.
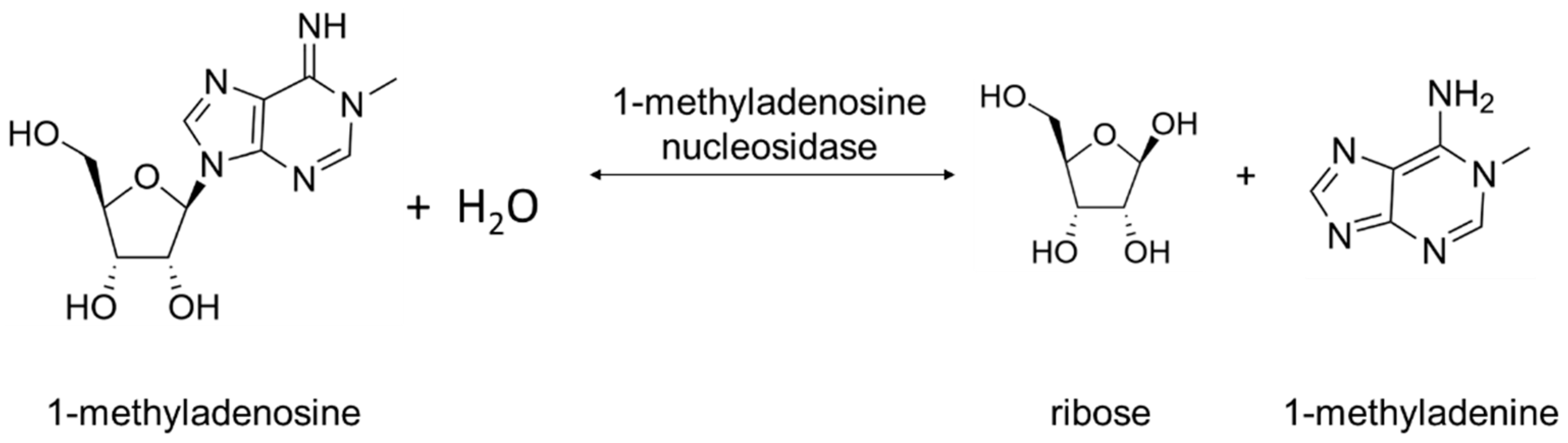
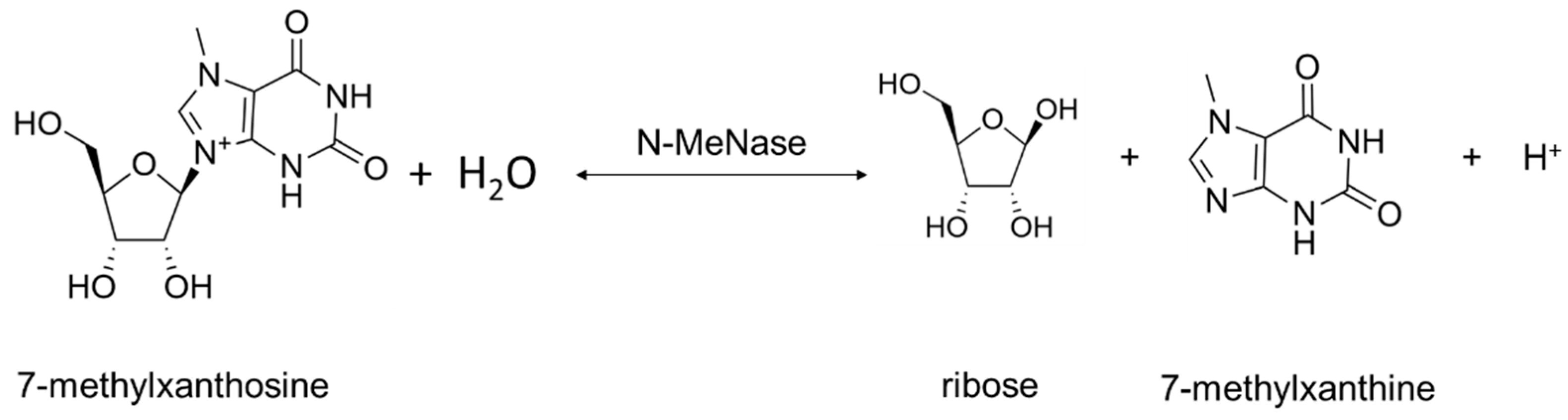
Two types of Rih hydrolases should be noted. These are 1-methyladenosine nucleosidase (EC 3.2.2.13) and N-methyl nucleosidase (7-methylxanthosine nucleosidase, N-MeNase, EC 3.2.2.25). Reactions catalyzed by them are shown in Figure 4 and Figure 5, respectively. The interesting thing about them is that both of these hydrolases can only catalyze the specific reaction below and cannot cleave any other purines [39][40][41][42][43]. The 1-methyladenosine nucleosidase has only been found in starfish [39][40][44]. It catalyzes the production of 1-methyladenine, which is responsible for inducing oocyte maturation and is found in ovaries of starfish. N-MeNase has been found in tea and coffee plants [41][42][43]. It catalyzes the production of 7-methylxanthine in these plants, which then turns into theobromine and caffeine. Thus, this nucleoside hydrolase also has an important role in the metabolism of the respective organism.
Figure 4. 1-Methyladenosine cleavage catalyzed by 1-methyladenosine nucleosidase.
Figure 5. 7-methylxanthosine cleavage catalyzed by N-MeNase.
From the information above, it can be concluded that for some organisms, Rih hydrolases do not play a key role in metabolism, while for others, they play one of the key roles in vital activity or under stress.
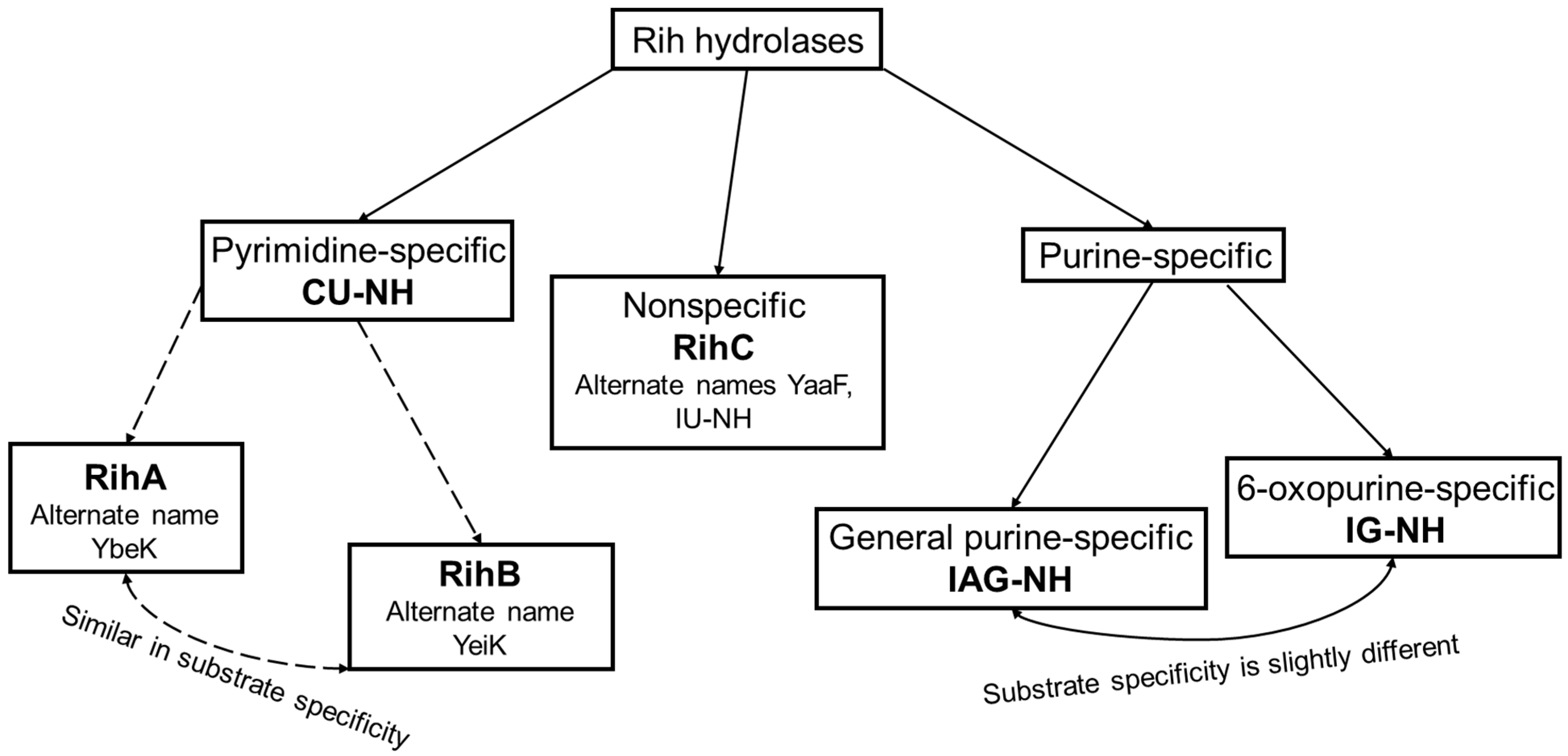
According to their substrate specificity, these hydrolases can be divided into three categories: pyrimidine-specific, purine-specific, and nonspecific (Figure 6) (excluding 1-methyladenosine nucleosidase and N-methyl nucleosidase since they catalyze highly specific reactions, although both could be considered purine-specific). Pyrimidine hydrolases (also known as CU-NH, cytidine–uridine-preferring nucleoside hydrolases) are specific to ribonucleosides containing a pyrimidine nitrogenous base (uracil, cytosine). Currently, hydrolases of this type are called RihA and RihB (alternative names: pyrimidine-specific NH, YbeK for RihA hydrolase, YeiK for RihB hydrolase), discovered in E. coli and first named so by researchers in [45]. There is not much difference between these two types of hydrolases. They have very similar substrate specificity, although it was noted by the researchers in [45] that while RihA hydrolase catalyzed only the cleavage of pyrimidine nucleosides, RihB also catalyzed the cleavage of purine nucleosides, albeit at an extremely low rate (much lower rate than nonspecific RihC from the same organism). An example of a reaction catalyzed by these types of hydrolases is shown in Figure 1. Hydrolases of this type are found mainly in bacteria [45][46][47][48][49]. Hydrolases of the second type are specific to ribonucleosides with purine nitrogenous bases (adenine, guanine, xanthine, hypoxanthine). These hydrolases can, in turn, be divided into two categories: specific to all purine bases (abbreviated as IAG-NH, i.e., inosine–adenosine–guanosine-preferring nucleoside hydrolase) or specific to 6-oxopurine bases (abbreviated as IG-NH, i.e., inosine–guanosine-preferring nucleoside hydrolase). An example of a reaction for this type of hydrolase is shown in Figure 2. Hydrolases of this type are found in archaea [50][51][52][53], protozoa [3][5][12][13][14], some bacteria [20], fungi [30][33], and insects [36]. Hydrolases of the third type are nonspecific purine–pyrimidine hydrolases. Alternative names also include RihC, nonspecific NH, IU-NH (inosine–uridine-preferring NH), YaaF. These hydrolases can catalyze the cleavage of both purine and pyrimidine ribonucleosides. Most preferably, these hydrolases react with uridine and inosine, but other nucleosides are also hydrolyzed at a certain rate and specificity. Examples of reactions are shown in Figure 1 and Figure 2. Hydrolases of this type are found in a wide variety of organisms: bacteria [21][34][45][54][55][56][57][58][59][60][61][62][63][64][65][66], plants [23][24][25][26][27], yeast [28][38], fungi [29][32], metazoa [34][35][37], and also some protozoa and archaea [53].
Figure 6. Types of Rih hydrolases.
3. Ribonucleoside Hydrolase Structure
3.1. Rih Amino Acid Sequences
The wide distribution of an enzyme and the broad substrate specificity can usually be explained by differences in structure. Sometimes, the replacement of just one amino acid residue in the catalytic center can lead to a dramatic change in the properties of the enzyme. In this regard, it is important to consider the structure of different Rih hydrolases in order to explain differences in substrate specificity and such a wide variety of organisms containing genes for these hydrolases.
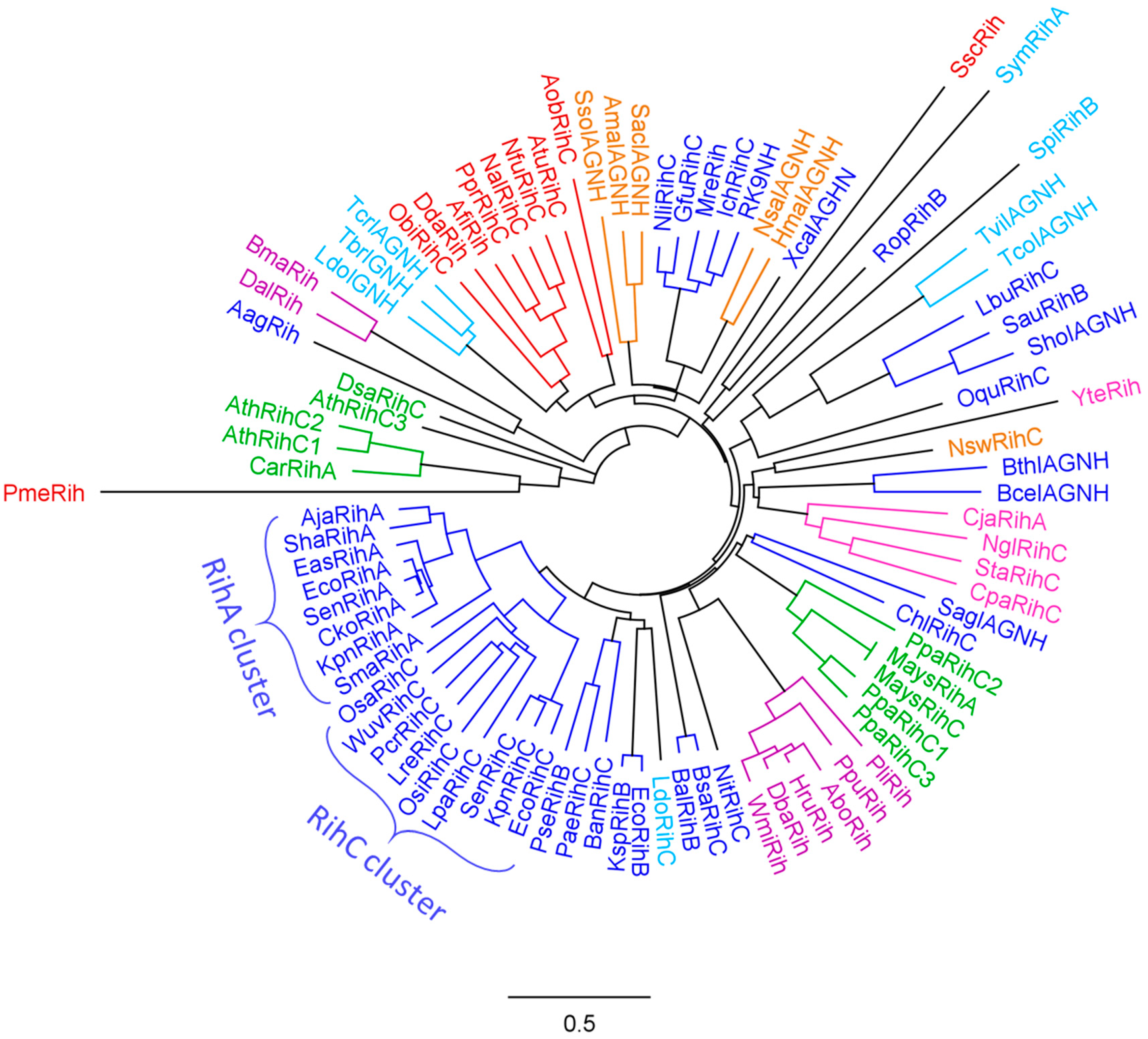
To assess the evolutionary relationship of various Rih hydrolases, a phylogenetic tree was constructed using the MUSCLE algorithm [67] and processed in the Geneious Prime program [68], as shown in Figure 7. A total of 88 hydrolase sequences from different organisms were selected. From closely related sequences, for example, from sequences of the same hydrolase from different strains of the same bacterial species, one was selected to increase sample diversity.
Figure 7. Phylogenetic tree of Rih hydrolases from various organisms. Enzymes from archaea are highlighted in orange, enzymes from bacteria are highlighted in blue, enzymes from fungi are highlighted in purple, enzymes from yeasts are highlighted in pink, enzymes from organisms belonging to metazoa are highlighted in red, from protozoan eukaryotes in light blue, and from plants in green.
On the phylogenetic tree (Figure 7), hydrolases from various organisms are marked with different colors. Some hydrolases are marked with asterisks in the table, and their name does not indicate the type of hydrolase. This was due to the fact that the type of hydrolase was not determined for these enzymes when they were made public. However, from the analysis of the phylogenetic tree and further from the alignment of amino acid sequences, it can be assumed that these hydrolases should belong to nonspecific RihC hydrolases.
Analyzing this phylogenetic tree, it is evident that hydrolases form several clusters according to the organisms in which they are found. There is a large cluster of bacterial Rih, and within this cluster one can see a small cluster of RihA hydrolases and a small cluster of RihC hydrolases, in which some RihB hydrolases are present in between. From the point of view of evolutionary development, this is logical: some organisms required hydrolases that act only on pyrimidine nucleosides, and all these RihA hydrolases are similar to each other; and some organisms needed different type of nucleosides, instead developing the ability to catalyze reactions with different nucleosides. RihB hydrolases are a more interesting subject for discussion. Despite the fact that they show the greatest efficiency in the catalysis of pyrimidine nucleosides, like RihA hydrolases, which is why they are combined with RihA hydrolases into one subtype, it was noted [45] that RihB hydrolases can also catalyze the cleavage reactions of some purine nucleosides at a very low rate. This can also be seen from the tree: they are evolutionarily closer to RihC hydrolases than to RihA hydrolases, being in between the former. Regarding bacterial Rih, it is also worth noting that among them there are some that are located far from the main bacterial cluster and are evolutionarily closer to other types of organisms. The reason for this diverse development of bacterial nucleoside hydrolases remains unclear.
Small clusters of other organisms are also clearly expressed on the phylogenetic tree: a separate cluster of animal hydrolases, a cluster of archaeal hydrolases, which are evolutionarily close to some bacterial hydrolases, and two separate unrelated clusters of yeast and fungal hydrolases. Some hydrolases from protozoa turned out to be evolutionarily close to the hydrolases of metazoa, while others turned out to be closer to bacterial ones. The most interesting on the evolutionary tree are Rih hydrolases from plants. It is known that many enzymes in plants are synthesized as several isozymes differing in primary structure and often in properties. The same is observed for hydrolases from plants Arabidopsis thaliana and moss Physcomitrella patens, each having three RihC isoenzymes. It is noteworthy that the hydrolases of these plants are in different clusters and that these clusters are evolutionarily distant from each other compared to other hydrolases. Why evolution has led to such a strong difference between one type of hydrolase in one type of organism remains not fully understood. This may be due to different ancestors of these plant types, and accordingly, different parent enzymes of current RihC hydrolases. In addition, it can be seen that some hydrolases are knocked out of their main clusters and often have elongated branches on the tree. The length of the branches indicates the average number of substitutions of amino acid residues in comparison with the neighbors, that is, in general, the proximity of the primary structure of specific hydrolases. Long branches indicate that although this hydrolase is close to other hydrolases nearby (has one tree node with them), it still differs significantly from them in its amino acid composition (the branch is elongated).
Three amino acid sequence alignments were constructed for the selected sequences. Proteins were grouped according to the type of substrate specificity: pyrimidine-specific (RihA and RihB), purine-specific (IAG-NH and IG-NH), and nonspecific (RihC). All available organisms for each protein type were selected for comparison.
The purine-specific hydrolases IAG-NH and IG-NH have been found in protozoa, archaea, and some bacteria. It can be seen that at the N-terminus of these enzymes, there is a strictly conserved DXDXXXDD sequence in the region of positions 10–20, which is responsible for the binding of calcium ion and ribose. Calcium binding is also aided by Asp residues at positions 205 and 305, while conserved GN and EXN sequences at positions 190 and 200, respectively, are involved in ribosyl ring binding [4][12][69]. The binding of the calcium ion and the ribose ring is a universal property of all ribonucleoside hydrolases. Therefore, the amino acid residues responsible for these functions are highly conserved, and this applies to all Rih hydrolases. As will be seen below, everything is much more complicated with the binding of a nitrogenous base, and accordingly the determination of amino acid residues that determine substrate specificity. In [12][14], it was suggested that residues W83 and W260 are responsible for the binding of the purine ring of purine nucleosides, explaining this by π-stacking interactions. However, in [4], studying 6-oxopurine-specific hydrolase, it is noted that there are no tryptophan residues in these structurally equivalent positions, although the enzyme is specific to inosine and guanosine. It was also noted in [69] which was about IAG-NH and not 6-oxopurine-specific hydrolase that these tryptophan residues are not preserved. In addition, the alignment also shows that although many purine-specific hydrolases contain semiconservative Phe, Tyr, and Trp residues (e.g., around positions 200, 204, 227), the positions considered in [12] and designated as ring binding of nitrogenous bases, are not conservative. It is possible that Trp, Tyr, or Phe residues are really involved in binding purines, but are located in other positions, or perhaps—even within this small group of hydrolases—each may have its own type of binding and stabilization of the purine ring of the ribonucleoside. The main features of the active site in this case are Asp residues that help bind calcium and ribose, Asn residues that are also involved in ribose binding, and a hydrophobic pocket in which the purine base should be located (possibly the conserved L(T/S)XXA sequence in the region of position 150 is involved in the formation of such a pocket, since it is formed from predominantly hydrophobic amino acid residues and does not have a strict described function in catalysis).
The pyrimidine-specific hydrolases RihA and RihB are found predominantly in bacteria, although these types of ribonucleoside hydrolases are also found in some other organisms, such as plants and protozoa. As in the case of purine hydrolases, these hydrolases have a highly conserved DXDXGXDD sequence at the N-terminus, which is necessary for binding the calcium ion and stabilizing ribose, which is part of the nucleosides [50]. Despite the fact that not only bacterial RihA and RihB were chosen for alignment but also enzymes from plants and yeast, it can be seen that the amino acid sequences of all the presented enzymes contain a large number of highly conserved and semiconserved regions. This may be due to the similarity of the substrates—uridine and cytidine—as well as the fact that there are only two of them. It can be assumed that catalysis proceeds in almost the same way regardless of the source of this type of enzyme. Presumably, highly conserved Asp and Asn residues are required for both calcium binding and ribose binding and stabilization of the transition state during catalysis. Hydrophobic amino acid residues, which are both conservative and semiconservative in these enzymes, are necessary for the formation of a hydrophobic pocket in the active site of the enzyme, among which the following residues are present: Leu, Ile, Val, Ala. In [50], the authors indicate that the binding of the pyrimidine ring in RihB (and accordingly, the same can be assumed about RihA due to the high similarity of these types of hydrolases) involves the Asn(Val/Ile)His sequence in the region of position 140, Phe in 238 position, Phe/Tyr in 311 position, His in 315 position, Tyr/Trp in 320 and His in 329 position. These residues are indeed conserved or semiconserved in RihA and RihB regardless of the source of the enzyme, and may aid in the binding of the nitrogenous base, which is the most hydrophobic part of the nucleoside.
Nonspecific RihC hydrolases have the widest distribution between all types of Rih hydrolases. These hydrolases have been found in a wide variety of organisms, ranging from bacteria to metazoan. For this type of hydrolase, as in the previous two cases, there is a highly conserved region DXDXXXDD in the region of the N-terminus (for some fungal hydrolases, this region is represented as DXDXXXXXDD), which is necessary for binding the calcium ion and the 2′-OH group of the ribose ring. In general, these hydrolases also contain the Asp and Asn amino acid residues that aid in ribose and calcium binding along the amino acid sequences and hydrophobic regions formed by conserved and semiconservative amino acid residues. It is more difficult to identify key residues in RihC that help to stabilize nitrogenous bases [62], suggesting that His and Tyr residues at positions 404 and 405 and His at position 428 are key to the stabilization of purine bases in RihC from E. coli. At the same time, Tyr400, Tyr405, and His428 were proposed in [70] for RihC as key residues binding the purine ring. The same catalytic triad was proposed for the mechanism of catalysis of pyrimidine nucleosides (to stabilize pyrimidine bases). However, paying attention to the alignment, one can clearly see that these residues are not conservative. For some RihC hydrolases, the second tyrosine residue from this triad is indeed retained, and the histidine residue is highly conserved for all RihC except fungal ones. It should be noted that the first residue of the triad does not have a strictly defined position in the amino acid sequence of hydrolases of this type, and in principle, it may not be a strictly conserved His or Tyr residue. However, it has been shown [9][15][16][62][70], that these amino acid residues are required for catalysis. Possibly, the low conservatism in their position can be explained by the difference in the structure of these hydrolases depending on the organism. Evolutionarily, these residues might have “migrated” along the amino acid sequence in one direction or another, depending on the organism. Moreover, the substrate specificity of RihC hydrolases, although generally similar (they can hydrolyze both purine and pyrimidine ribonucleosides), still differs depending on the organism, which may be due precisely to the position of this triad and in turn can be explained by the particular organism in which this RihC hydrolase is produced and what needs have been formed in this organism in the course of evolution in relation to nitrogenous bases and nucleosides.
3.2. Rih Crystal Structure
To date, a number of crystal structures of the Rih family enzymes of different types have been obtained. The researchers managed to obtain crystals of recombinant ribonucleoside hydrolases from different organisms: the structures of enzymes from bacteria, archaea, protozoa, and even plants and metazoa are known.
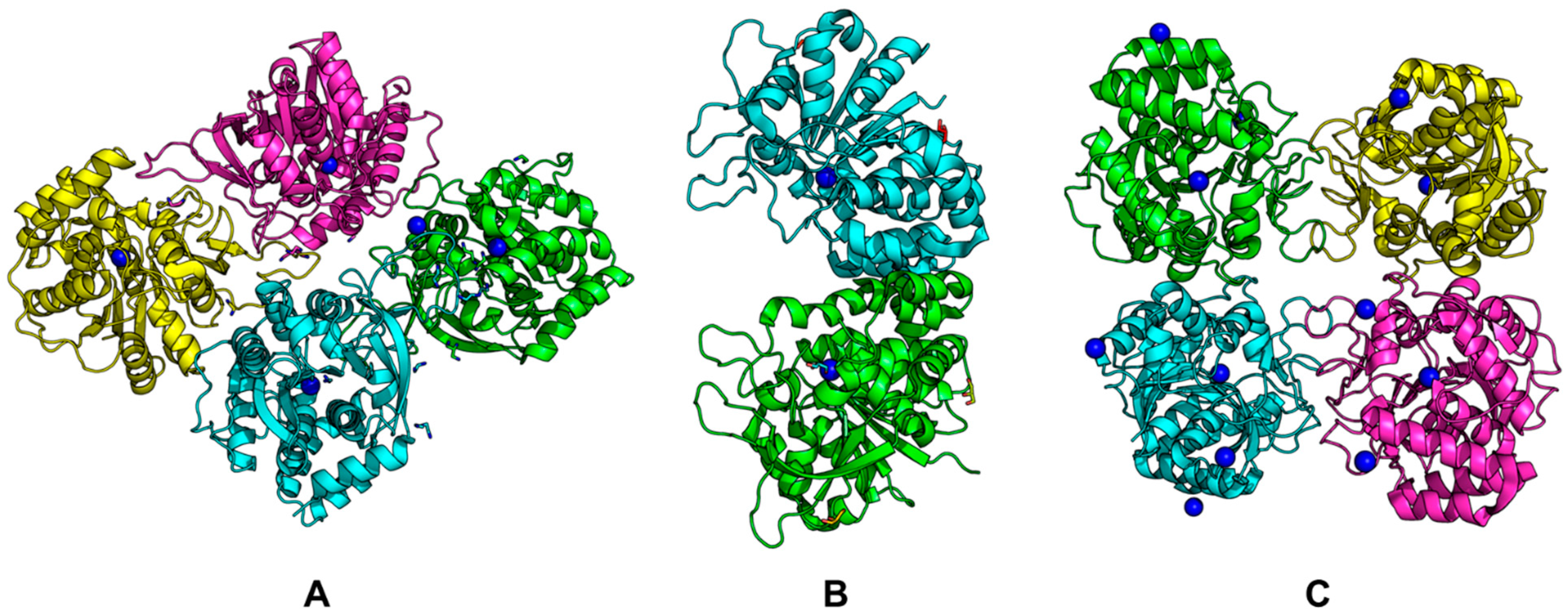
Examples of structures are shown in Figure 8 (PDB codes for each structure are in parentheses). It can be seen that Rih hydrolases can be a tetramer (Figure 8A,C) or a dimer (Figure 8B), and the structures can be both symmetric and asymmetric.
Figure 8. Rih crystal structures. (A) RihA from Zea mays (6ZK1), (B) IAG-NH from Trypanosoma vivax (1HOZ), (C) RihB from Gardnerella vaginalis (6BA0). Calcium ions are shown in blue.
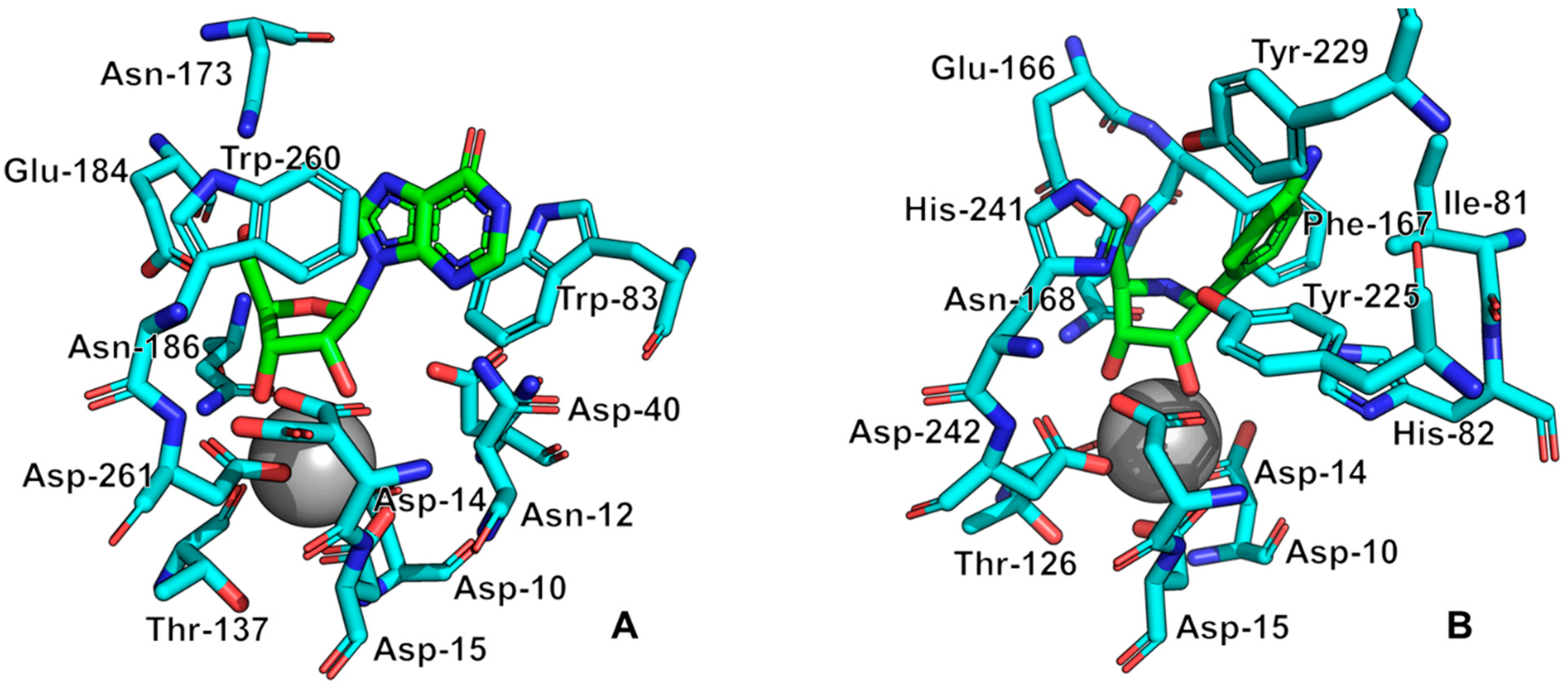
All currently known three-dimensional structures of Rih hydrolases contain at least one calcium ion per subunit, which is essential for catalysis [8][15][16][34][70][71][72]. At the same time, some structures have two (Figure 8A) or even three and four (Figure 8C) calcium ions in them. These additional calcium ions may help stabilize the structure itself and not play any role in the catalysis. Figure 9 shows fragments of the active site of IAG-NH from Trypanosoma vivax in complex with inosine (Figure 9A) and RihC from Crithidia fasciculata in complex with pAPIR (para-aminophenyliminoribitol; Figure 9B). For the IAG-NH enzyme in Figure 9A, one can observe the close arrangement of two tryptophan residues, which, as described earlier, are involved in π-stacking and thus stabilize the nitrogenous base of the nucleoside. Figure 9B shows the close proximity of the histidine and tyrosine residues of the RihC enzyme to the nitrogenous base in the nucleoside (in this case pAPIR plays the role of the nucleoside), which indicates their importance in substrate binding [73]. Both enzymes show the presence of conserved Asp, Asn, Glu, and Thr residues involved in the binding of the calcium ion or the nucleoside ribose ring.
Figure 9. Active site fragments of IAG-NH from T. vivax in complex with inosine ((A), PDB ID: 1KIC) and RihC from C. fasciculata in complex with pAPIR ((B), PDB ID: 2MAS). Molecules in the complex with the enzyme are indicated in green, key enzyme amino acid residues are indicated in cyan, calcium ions are indicated by a gray sphere. Key active site residues are numbered for each case.
The mechanism of catalysis for purine Rih hydrolases generally corresponds to the mechanism of SN1 nucleophilic substitution with the formation of an oxocarbenium ion in the transition state [11][74]. In this transition state, the N-glycosidic bond of the nucleoside is in a nearly broken state with an interatomic distance of 2Å, while the attacking water molecule is at a distance of approximately 3Å. The study of kinetic isotope effects also showed that protonation of the N7 atom of the purine ring of the nucleoside precedes the appearance of the transition state and leads to destabilization of the N-glycosidic bond. Based on this, three main points in the hydrolysis of purine nucleosides can be distinguished: steric and electrostatic stabilization of the oxocarbenium ion, activation of the nucleophilic water molecule, and activation of the leaving group.
The activation of the water molecule occurs due to its noncovalent binding to the calcium ion in the active center and the pulling of the proton of this water molecule towards itself by the Asp residue. In the noncatalytic solvolysis of nucleosides, it was found that the free OH groups of ribose have a stabilizing effect on the N-glycosidic bond and prevent the formation of a positively charged oxocarbenium ion [75]. For enzymatic catalysis, however, the presence of free OH groups is essential. Presumably, the oxocarbenium transition state is significantly stabilized by the interactions of the metalloenzyme and ribose in two ways. The first is the accumulation of binding energy, which is used to bend the conformation of the ribose ring and bring it into an activated state (steric catalysis). The second is the polarization of some hydroxyl groups with the subsequent appearance of (partial) negative charges on them, which stabilize the resulting positive oxocarbenium ion (electrostatic catalysis).
As noted earlier, the appearance of the transition state is preceded by the protonation of the N7 atom of the purine ring of the nucleoside. During this process, a good leaving group appears, facilitating the destruction of the N-glycosidic bond. The mechanism of such stabilization remains not completely understood, since the Trp amino acid residues are in the immediate environment of the nitrogenous base in the enzyme, but the indole ring of the tryptophan residue cannot be a classical acid that activates the N7 atom, since this ring is not ionized. It was suggested [14] that due to π-stacking, the pKa of the N7 nitrogenous base atom increases, due to which this atom is protonated by water.
In the case of catalysis by pyrimidine or nonspecific hydrolases, the general mechanism is similar to the previous case. Since the calcium ion and conservative aspartic acid residues are present in all enzymes, the activation of the nucleophilic water molecule and the stabilization of the oxocarbenium ion occur in the same way as described above. The activation of the leaving group itself also occurs due to the protonation of the N3 atom of the pyrimidine (analogous to the N7 atom of the purine) nitrogenous base; however, in this case, tyrosine and histidine residues are located near the ring of the nitrogenous base, presumably involved in protonation and the formation of a good leaving group. It is assumed that for RihC hydrolases, during the catalysis of pyrimidine nucleoside cleavage, the protonation of the N3 atom occurs via the histidine residue itself, while in the catalysis of the cleavage of purine nucleosides, the protonation of the N7 atom occurs along the His-Tyr-Tyr chain. At the same time, in the pyrimidine hydrolases RihA and RihB, tyrosine residues are nonconservative; therefore, these hydrolases either cannot catalyze the cleavage of purine nucleosides at all, or they can, but are orders of magnitude worse than the cleavage of pyrimidine nucleosides.
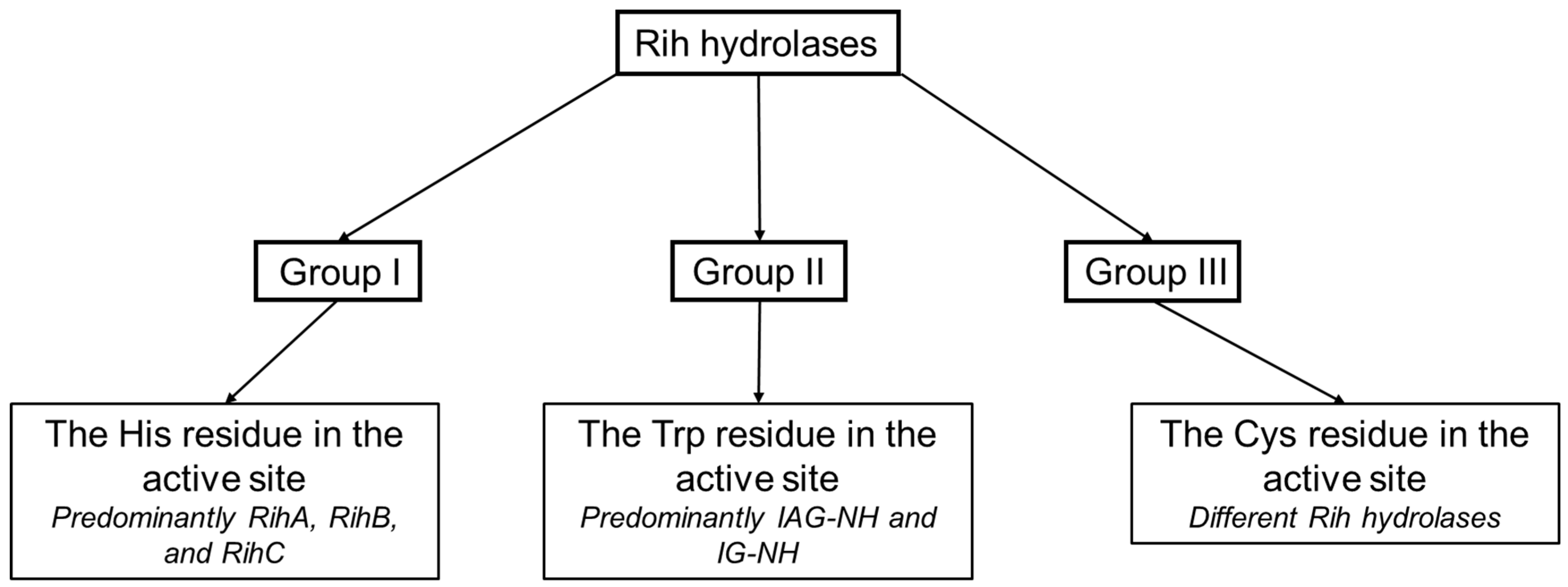
Based on the structure of the active center, namely, depending on the key amino acid residues stabilizing the nitrogenous base of the nucleoside, a classification of Rih nucleoside hydrolases into three groups not based on substrate specificity was proposed (Figure 10). It is customary to include hydrolases in group I that have a histidine residue in their active center (as well as tyrosine residues conjugated with this residue involved in the protonation of the nitrogenous base ring), in group II hydrolases that have a tryptophan residue instead of this histidine residue (and conjugate with it the second tryptophan residue at a distance, forming π-stacking interactions with the purine ring), and in group III hydrolases with a cysteine residue in this position (which usually also has a paired cysteine residue at a distance, also involved in catalysis).
Figure 10. Schematic dividing of Rih hydrolases into three groups depending on the catalytically important residues in the active site.
Hydrolases of the first two groups are currently the most studied and most common. In these two groups, there is also a fairly clear division of hydrolases according to their substrate specificity. The third group includes hydrolases with different substrate preferences. Despite the fact that the cysteine residue in the active site is more similar in function to the group I histidine residue and that the protonation of the nitrogenous base occurs in a similar way to the hydrolases of the first group, purine-specific hydrolases have also been found in this group (for example, in [35]).
It should be noted that this classification cannot be considered final, since some Rih hydrolases do not contain any of the described amino acid residues in this catalytically important position, but contain a proline residue. This can be seen in the amino acid alignments described in the previous section for some of the IAG-NH and RihC hydrolases. The presence of a proline residue at this position is not a single mutation associated with the evolutionary development of a single enzyme, but is observed for a number of Rih hydrolases from both bacteria and archaea. If for RihC hydrolases from fungi, which have a proline residue in the position binding the nitrogenous ring, a histidine residue is located next to this proline residue, i.e., these changes most likely do not affect catalysis, then for purine-specific hydrolases, there is neither a histidine residue nor a tryptophan residue in the immediate vicinity of this proline residue, which should bind the nitrogenous base in the active center. “Proline” hydrolases have currently not been studied, so the exact mechanism of activation of the leaving group cannot be explained; however, the researchers assume that the mechanism is similar to the activation mechanism in group II: the proline residue increases the pKa of the nitrogenous base and facilitates the protonation of nitrogen by water, which creates a good leaving group. However, to accurately establish the mechanism of catalysis by these hydrolases, experiments and analysis of kinetic data and structure are required.
References
- ChemDraw PerkinElmer JS. Available online: https://chemdrawdirect.perkinelmer.cloud/js/sample/index.html# (accessed on 28 August 2023).
- Miller, R.L.; Sabourin, C.L.K.; Krenitsky, T.A. Nucleoside Hydrolases from Trypanosoma Cruzi. J. Biol. Chem. 1984, 259, 5073–5077.
- Nolan, L.L.; Kidder, G.W. Inhibition of Growth and Purine-Metabolizing Enzymes of Trypanosomid Flagellates by N6-Methyladenine. Antimicrob. Agents Chemother. 1980, 17, 567–571.
- Vandemeulebroucke, A.; Minici, C.; Bruno, I.; Muzzolini, L.; Tornaghi, P.; Parkin, D.W.; Versées, W.; Steyaert, J.; Degano, M. Structure and Mechanism of the 6-Oxopurine Nucleosidase from Trypanosoma brucei brucei. Biochemistry 2010, 49, 8999–9010.
- Parkin, D.W.; Horenstein, B.A.; Abdulah, D.R.; Estupinan, B.; Schramm, V.L. Nucleoside Hydrolase from Crithidia Fasciculuta. Metabolic Role, Purification, Specificity, and Kinetic Mechanism. J. Biol. Chem. 1991, 266, 20658–20665.
- Suthar, N.; Goyal, A.; Dubey, V.K. Identification of Potential Drug Targets of Leishmania Infantum by In-Silico Genome Analysis. Lett. Drug Des. Discov. 2009, 6, 620–622.
- Sales, E.M.; Sousa, G.S.; Belouezzane, C.; Almeida, F.C.L.; Figueroa-Villar, J.D. Expression, Purification and Spectrophotometric Analysis of Nucleoside Hydrolase from Leishmania chagasi (LcNH). Protein Expr. Purif. 2019, 161, 40–48.
- Shi, W.; Schramm, V.L.; Almo, S.C. Nucleoside Hydrolase from Leishmania Major. Cloning, Expression, Catalytic Properties, Transition State Inhibitors, and the 2.5-Å Crystal Structure. J. Biol. Chem. 1999, 274, 21114–21120.
- Degano, M.; Almo, S.C.; Sacchettini, J.C.; Schramm, V.L. Trypanosomal Nucleoside Hydrolase. A Novel Mechanism from the Structure with a Transition-State Inhibitor. Biochemistry 1998, 37, 6277–6285.
- Fish, W.R.; Looker, D.L.; Marr, J.J.; Berens, R.L. Purine Metabolism in the Bloodstream Forms of Trypanosoma gambiense and Trypanosoma rhodesiense. Biochim. Biophys. Acta 1982, 719, 223–231.
- Horenstein, B.A.; Parkin, D.W.; Estupifiiin, B.; Schramm, V.L. Transition-State Analysis of Nucleoside Hydrolase from Crithidia fasciculata. Biochemistry 1991, 30, 10788–10795.
- Versées, W.; Barlow, J.; Steyaert, J. Transition-State Complex of the Purine-Specific Nucleoside Hydrolase of T. vivax: Enzyme Conformational Changes and Implications for Catalysis. J. Mol. Biol. 2006, 359, 331–346.
- Versées, W.; Decanniere, K.; Pellé, R.; Depoorter, J.; Brosens, E.; Parkin, D.W.; Steyaert, J. Structure and Function of a Novel Purine Specific Nucleoside Hydrolase from Trypanosoma vivax. J. Mol. Biol. 2001, 307, 1363–1379.
- Versées, W.; Decanniere, K.; Van Holsbeke, E.; Devroede, N.; Steyaert, J. Enzyme-Substrate Interactions in the Purine-Specific Nucleoside Hydrolase from Trypanosoma vivax. J. Biol. Chem. 2002, 277, 15938–15946.
- Gopaul, D.N.; Meyer, S.L.; Degano, M.; Sacchettini, J.C.; Schramm, V.L. Inosine−Uridine Nucleoside Hydrolase from Crithidia fasciculata. Genetic Characterization, Crystallization, and Identification of Histidine 241 as a Catalytic Site Residue. Biochemistry 1996, 35, 5963–5970.
- Degano, M.; Gopaul, D.N.; Scapin, G.; Schramm, V.L.; Sacchettini, J.C. Three-Dimensional Structure of the Inosine−Uridine Nucleoside N-Ribohydrolase from Crithidia fasciculata. Biochemistry 1996, 35, 5971–5981.
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry: International Edition, 8th ed.; W. H. Freeman & Co., Ltd.: New York, NY, USA, 2021; ISBN 1319381499.
- Bzowska, A.; Kulikowska, E.; Shugar, D. Purine Nucleoside Phosphorylases: Properties, Functions, and Clinical Aspects. Pharmacol. Ther. 2000, 88, 349–425.
- Bzowska, A. Purine and Pyrimidine Nucleoside Phosphorylases—Remarkable Enzymes Still Not Fully Understood. Postep. Biochem. 2015, 61, 260–273.
- Liang, L.; He, X.; Liu, G.; Tan, H. The Role of a Purine-Specific Nucleoside Hydrolase in Spore Germination of Bacillus thuringiensis. Microbiology 2008, 154, 1333–1340.
- Todd, S.J.; Moir, A.J.G.; Johnson, M.J.; Moir, A. Genes of Bacillus cereus and Bacillus anthracis Encoding Proteins of the Exosporium. J. Bacteriol. 2003, 185, 3373–3378.
- Guimarães, A.P.; Oliveira, A.A.; Da Cunha, E.F.F.; Ramalho, T.C.; França, T.C.C. Analysis of Bacillus anthracis Nucleoside Hydrolase via in Silico Docking with Inhibitors and Molecular Dynamics Simulation. J. Mol. Model. 2011, 17, 2939–2951.
- Kopečná, M.; Blaschke, H.; Kopečný, D.; Vigouroux, A.; Končitíková, R.; Novák, O.; Kotland, O.; Strnad, M.; Moréra, S.; von Schwartzenberg, K. Structure and Function of Nucleoside Hydrolases from Physcomitrella patens and Maize Catalyzing the Hydrolysis of Purine, Pyrimidine, and Cytokinin Ribosides. Plant Physiol. 2013, 163, 1568–1583.
- Stasolla, C.; Katahira, R.; Thorpe, T.A.; Ashihara, H. Purine and Pyrimidine Nucleotide Metabolism in Higher Plants. J. Plant Physiol. 2003, 160, 1271–1295.
- Zrenner, R.; Stitt, M.; Sonnewald, U.; Boldt, R. Pyrimidine and Purine Biosynthesis and Degradation in Plants. Annu. Rev. Plant Biol. 2006, 57, 805–836.
- Thicklin, L.; Shamsuddin, A.; Alahmry, F.; Gezley, C.; Brown, E.; Stone, J.; Burns-Carver, E.; Kline, P.C. Purification of a Non-Specific Nucleoside Hydrolase from Alaska Pea Seeds. Protein Expr. Purif. 2019, 154, 140–146.
- Riegler, H.; Geserick, C.; Zrenner, R. Arabidopsis thaliana Nucleosidase Mutants Provide New Insights into Nucleoside Degradation. New Phytol. 2011, 191, 349–359.
- Kurtz, J.E.; Exinger, F.; Erbs, P.; Jund, R. The URH1 Uridine Ribohydrolase of Saccharomyces cerevisiae. Curr. Genet. 2002, 41, 132–141.
- Rizzatti, A.C.S.; Sandrim, V.C.; Jorge, J.A.; Terenzi, H.F.; Polizeli, M.D.L.T.M. Influence of Temperature on the Properties of the Xylanolytic Enzymes of the Thermotolerant Fungus Aspergillus phoenicis. J. Ind. Microbiol. Biotechnol. 2004, 31, 88–93.
- Elshafei, A.M.; Mohamed, L.A.; Hassan, M.M. Degradation of Purine Ribonucleosides by Extracts of Penicillium viridicatum. Afr. J. Microbiol. Res. 2011, 5, 316–323.
- Elshafei, A.M.; Abu-Shady, M.R.; El-Beih, F.M.; Mohamed, L.A. Mode and Extent of Degradation of Adenosine and Guanosine by Extracts of Aspergillus terricola. Microbiol. Res. 1995, 150, 291–295.
- Allam, A.M.; Hassan, M.M.; Ghanem, B.S.; Elzainy, T.A. Nature of the Enzymes That Catalyse the Cleavage of the N-Glycosidic Bond of Pyrimidine Ribonucleosides in Some Filamentous Fungi. Biochem. Syst. Ecol. 1987, 15, 515–517.
- Abdel-Fatah, O.M.; Elsayed, M.A.; Elshafei, A.M. Hydrolytic Cleavage of Purine Ribonucleosides in Aspergillus phoenicis. J. Basic. Microbiol. 2003, 43, 439–448.
- Versées, W.V.; Van Holsbeke, E.; De Vos, S.; Decanniere, K.; Zegers, I.; Steyaert, J. Cloning, Preliminary Characterization and Crystallization of Nucleoside Hydrolases from Caenorhabditis elegans and Campylobacter jejuni. Acta Cryst. 2003, 59, 1087–1089.
- Singh, R.K.; Steyaert, J.; Versées, W. Structural and Biochemical Characterization of the Nucleoside Hydrolase from C. elegans Reveals the Role of Two Active Site Cysteine Residues in Catalysis. Protein Sci. 2017, 26, 985–996.
- Ribeiro, J.M.C.; Valenzuela, J.G. The Salivary Purine Nucleosidase of the Mosquito, Aedes aegypti. Insect Biochem. Mol. Biol. 2003, 33, 13–22.
- Tarr, H.L.A. Fish Muscle Riboside Hydrolases. J. Biol. Chem. 1955, 127, 386–391.
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide Riboside Promotes Sir2 Silencing and Extends Lifespan via Nrk and Urh1/Pnp1/Meu1 Pathways to NAD+. Cell 2007, 129, 473–484.
- Shirai, H.; Kanatani, H. 1-Methyladenosine Ribohydrolase in the Starfish Ovary and Its Relation to Oocyte Maturation. Exp. Cell Res. 1972, 75, 79–88.
- Tarr, H.L.A. 1-Methyladenosine Hydrolase of Starfish (Pisaster ochraceous). J. Fish. Res. Board. Can. 1973, 30, 1861–1866.
- Roberts, M.F.; Wallert, G.R. N-Methyltransferases and 7-Methyl-N9-Nucleoside Hydrolase Activity in Coffea arabica and the Biosynthesis of Caffeine. Phytochemistry 1979, 18, 451–455.
- Waller, G.R.; MacVean, C.D.; Suzuki, T. High Production of Caffeine and Related Enzyme Activities in Callus Cultures of Coffea arabica L. Plant Cell Rep. 1983, 2, 109–112.
- Negishi, O.; Ozawa, T.; Imagawa, H. N-Methyl Nucleosidase from Tea Leaves. Agric. Biol. Chem. 1988, 52, 169–175.
- Kanatani, H.; Shirai, H. Chemical Structural Requirements for Induction of Oocyte Maturation and Spawning in Stareishes. Dev. Growth Differ. 1971, 13, 53–64.
- Petersen, C.; Møller, L.B. The RihA, RihB, and RihC Ribonucleoside Hydrolases of Escherichia coli. Substrate Specificity, Gene Expression, and Regulation. J. Biol. Chem. 2001, 276, 884–894.
- Garau, G.; Muzzolini, L.; Tornaghi, P.; Degano, M. Active Site Plasticity Revealed from the Structure of the Enterobacterial N-Ribohydrolase RihA Bound to a Competitive Inhibitor. BMC Struct. Biol. 2010, 10, 14.
- West, T.P. Degradation of Pyrimidine Ribonucleosides by Pseudomonas aeruginosa. Antonie van Leeuwenhoek 1996, 69, 331–335.
- West, T.P. Pyrimidine Ribonucleoside Catabolic Enzyme Activities of Pseudomonas pickettii. Antonie van Leeuwenhoek 1994, 66, 307.
- West, T.P. Pyrimidine Base and Ribonucleoside Catabolic Enzyme Activities of the Pseudomonas diminuta Group. FEMS Microbiol. Lett. 1992, 99, 305–310.
- Porcelli, M.; Concilio, L.; Peluso, I.; Marabotti, A.; Facchiano, A.; Cacciapuoti, G. Pyrimidine-Specific Ribonucleoside Hydrolase from the Archaeon Sulfolobus solfataricus—Biochemical Characterization and Homology Modeling. FEBS J. 2008, 275, 1900–1914.
- Porcelli, M.; De Leo, E.; Del Vecchio, P.; Fuccio, F.; Cacciapuoti, G. Thermal Unfolding of Nucleoside Hydrolases from the Hyperthermophilic Archaeon Sulfolobus solfataricus: Role of Disulfide Bonds. Protein Pept. Lett. 2012, 19, 369–374.
- Porcelli, M.; De Leo, E.; Marabotti, A.; Cacciapuoti, G. Site-Directed Mutagenesis Gives Insights into Substrate Specificity of Sulfolobus solfataricus Purine-Specific Nucleoside Hydrolase. Ann. Microbiol. 2012, 62, 881–887.
- Minici, C.; Cacciapuoti, G.; De Leo, E.; Porcelli, M.; Degano, M. New Determinants in the Catalytic Mechanism of Nucleoside Hydrolases from the Structures of Two Isozymes from Sulfolobus solfataricus. Biochemistry 2012, 51, 4590–4599.
- Terada, M.; Tatibana, M.; Hayaishi, O. Purification and Properties of Nucleoside Hydrolase from Pseudomonas fluorescens. J. Biol. Chem. 1967, 242, 5578–5585.
- Ueda, A.; Attila, C.; Whiteley, M.; Wood, T.K. Uracil Influences Quorum Sensing and Biofilm Formation in Pseudomonas aeruginosa and Fluorouracil Is an Antagonist. Microb. Biotechnol. 2009, 2, 62–74.
- Savinova, O.S.; Glazunova, O.A.; Moiseenko, K.V.; Begunova, A.V.; Rozhkova, I.V.; Fedorova, T.V. Exoproteome Analysis of Antagonistic Interactions between the Probiotic Bacteria Limosilactobacillus reuteri Lr1 and Lacticaseibacillus rhamnosus f and Multidrug Resistant Strain of Klebsiella pneumonia. Int. J. Mol. Sci. 2021, 22, 999.
- Khoroshkin, M.S.; Leyn, S.A.; Van Sinderen, D.; Rodionov, D.A. Transcriptional Regulation of Carbohydrate Utilization Pathways in the Bifidobacterium Genus. Front. Microbiol. 2016, 7, 120.
- Mitsukawa, Y.; Hibi, M.; Matsutani, N.; Horinouchi, N.; Takahashi, S.; Ogawa, J. New Nucleoside Hydrolase with Transribosylation Activity from Agromyces sp. MM-1 and Its Application for Enzymatic Synthesis of 2′-O-Methylribonucleosides. J. Biosci. Bioeng. 2018, 125, 38–45.
- Mitsukawa, Y.; Hibi, M.; Matsutani, N.; Horinouchi, N.; Takahashi, S.; Ogawa, J. A Novel Nucleoside Hydrolase from Lactobacillus buchneri LBK78 Catalyzing Hydrolysis of 2′-O-Methylribonucleosides. Biosci. Biotechnol. Biochem. 2016, 80, 1568–1576.
- Kim, H.S.; Lee, J.H.; Lee, W.S.; Bang, W.G. Genes Encoding Ribonucleoside Hydrolase 1 and 2 from Corynebacterium ammoniagenes. Microbiology 2006, 152, 1169–1177.
- Johansson, P.; Paulin, L.; Säde, E.; Salovuori, N.; Alatalo, E.R.; Björkroth, K.J.; Auvinen, P. Genome Sequence of a Food Spoilage Lactic Acid Bacterium, Leuconostoc gasicomitatum LMG 18811T, in Association with Specific Spoilage Reactions. Appl. Environ. Microbiol. 2011, 77, 4344–4351.
- Hunt, C.; Gillani, N.; Farone, A.; Rezaei, M.; Kline, P.C. Kinetic Isotope Effects of Nucleoside Hydrolase from Escherichia coli. Biochim. Biophys. Acta-Proteins Proteom. 2005, 1751, 140–149.
- Hansen, M.R.; Dandanell, G. Purification and Characterization of RihC, a Xanthosine-Inosine-Uridine- Adenosine-Preferring Hydrolase from Salmonella enterica Serovar Typhimurium. Biochim. Biophys. Acta-Gen. Subj. 2005, 1723, 55–62.
- Aučynaite, A.; Rutkiene, R.; Tauraite, D.; Meškys, R.; Urbonavičius, J. Identification of a 2′-O-Methyluridine Nucleoside Hydrolase Using the Metagenomic Libraries. Molecules 2018, 23, 2904.
- Arivett, B.; Farone, M.; Masiragani, R.; Burden, A.; Judge, S.; Osinloye, A.; Minici, C.; Degano, M.; Robinson, M.; Kline, P. Characterization of Inosine-Uridine Nucleoside Hydrolase (RihC) from Escherichia coli. Biochim. Biophys. Acta-Proteins Proteom. 2014, 1844, 656–662.
- Arevalo-Ferro, C.; Hentzer, M.; Reil, G.; Görg, A.; Kjelleberg, S.; Givskov, M.; Riedel, K.; Eberl, L. Identification of Quorum-Sensing Regulated Proteins in the Opportunistic Pathogen Pseudomonas aeruginosa by Proteomics. Environ. Microbiol. 2003, 5, 1350–1369.
- Multiple Sequence Alignment. Available online: https://www.ebi.ac.uk/Tools/msa/muscle/ (accessed on 10 August 2023).
- Geneious Prime. Available online: http://www.geneious.com/ (accessed on 10 August 2023).
- Porcelli, M.; Peluso, I.; Marabotti, A.; Facchiano, A.; Cacciapuoti, G. Biochemical Characterization and Homology Modeling of a Purine-Specific Ribonucleoside Hydrolase from the Archaeon Sulfolobus solfataricus: Insights into Mechanisms of Protein Stabilization. Arch. Biochem. Biophys. 2009, 483, 55–65.
- Iovane, E.; Giabbai, B.; Muzzolini, L.; Matafora, V.; Fornili, A.; Minici, C.; Giannese, F.; Degano, M. Structural Basis for Substrate Specificity in Group I Nucleoside Hydrolases. Biochemistry 2008, 47, 4418–4426.
- Fornili, A.; Giabbai, B.; Garau, G.; Degano, M. Energy Landscapes Associated with Macromolecular Conformational Changes from Endpoint Structures. J. Am. Chem. Soc. 2010, 132, 17570–17577.
- Giabbai, B.; Degano, M. Crystal Structure to 1.7 Å of the Escherichia coli Pyrimidine Nucleoside Hydrolase YeiK, a Novel Candidate for Cancer Gene Therapy. Structure 2004, 12, 739–749.
- Farone, A.; Farone, M.; Kline, P.; Quinn, T.; Sinkala, Z. A Practical Approach for Computing the Active Site of the Ribonucleoside Hydrolase of E. coli Encoded by RihC. Adv. Exp. Med. Biol. 2010, 680, 437–443.
- Horenstein, B.A.; Schramm, V.L. Electronic Nature of the Transition State for Nucleoside Hydrolase. A Blueprint for Inhibitor Design. Biochemistry 1993, 32, 7089–7097.
- Garrett, E.R.; Mehta, P.J. Solvolysis of Adenine Nucleosides. I. Effects of Sugars and Adenine Substituents on Acid Solvolyses. J. Am. Chem. Soc. 1972, 94, 8532–8541.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
20 Sep 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No