 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | IRAM IQBAL | -- | 4604 | 2023-09-16 00:10:08 | | | |
| 2 | Peter Tang | Meta information modification | 4604 | 2023-09-18 04:51:38 | | |
Video Upload Options
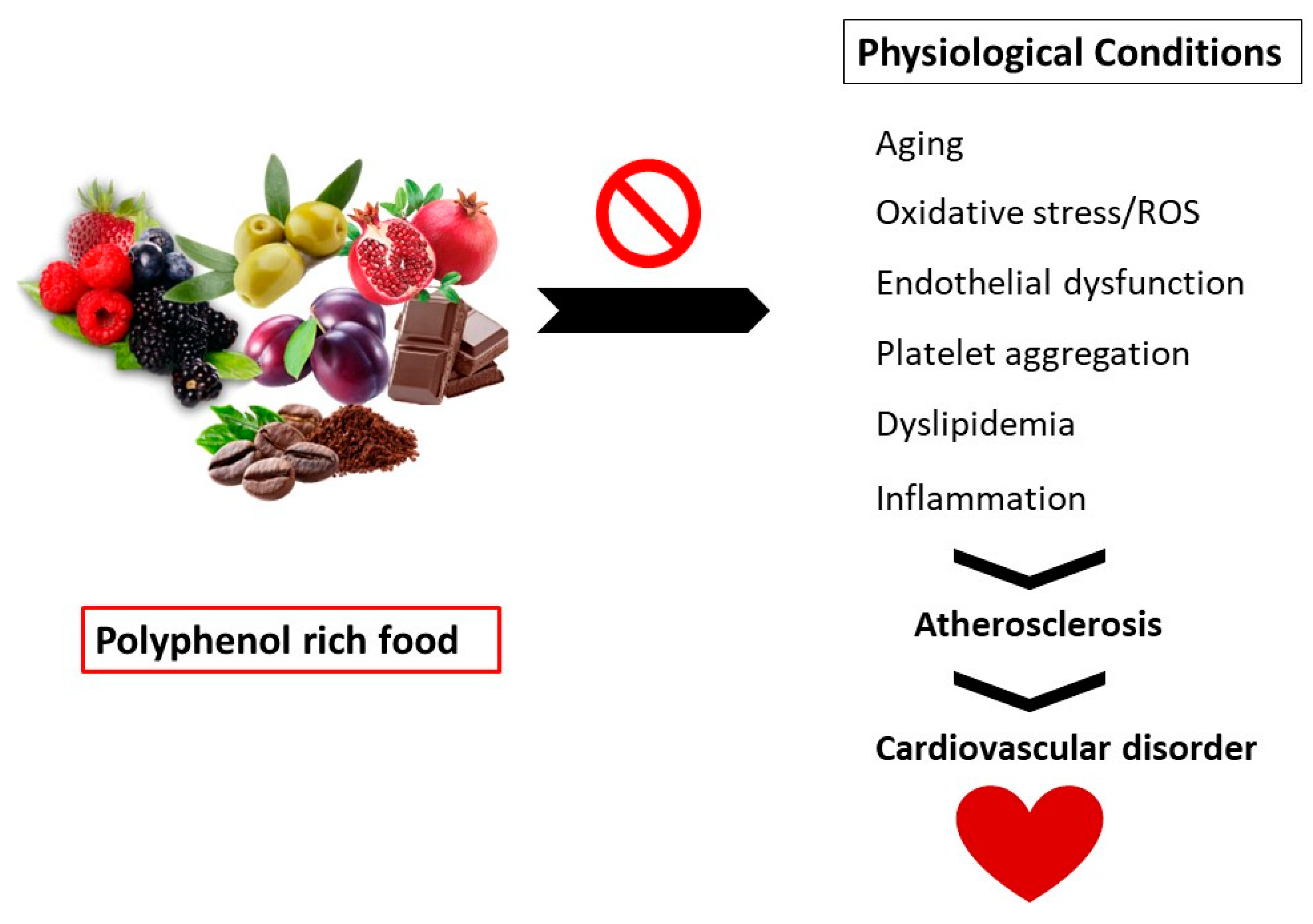
Polyphenols are secondary metabolites found in vegetables, fruits, and grains. These compounds exhibit several health benefits such as immune modulators, vasodilators, and antioxidants.
1. Introduction
2. Polyphenols
|
Plant Food |
Latin Name |
Edible Part |
Concentration mg/100 g |
Major Polyphenols |
References |
|---|---|---|---|---|---|
|
Apple |
Malus domestica |
Peel |
50–120 y |
Phlorizin, quercetin, phenolic acids (chlorogenic acid) |
|
|
Flesh |
0.2–0.9 |
||||
|
Total |
5–50 |
||||
|
Blackberry |
Rubus fruticosus |
Whole |
130–405 |
Anthocyanins, flavanols (EC), phenolic acid (ellagic acid) |
[20] |
|
Blueberry |
Vaccinium corymbosum |
Whole |
160–480 |
Anthocyanins, flavonols (quercetin), phenolic acids (chlorogenic acid) |
[20] |
|
Coffee |
Coffea arabica |
Beverage, filtered |
90 |
Phenolic acids (chlorogenic acid) |
[20] |
|
Chestnut (raw) |
Castanea sativa |
Whole nut |
547–1960 |
Hydroxybenzoic acids (gallic acid, ellagic acid), tannins |
[20] |
|
Cacao |
Theabroma cacao |
Beans, powder |
300–1100 x |
Flavanols (EC) |
[20] |
|
Green tea |
Camellia sinensis |
Extract |
29–103 x |
Flavanols (EC, EGCG) |
[20] |
|
Grapefruit |
Citrus x paradisi |
Flesh |
15–115 |
Flavonoids, phenolic acids |
[20] |
|
Olive oil, extra virgin |
Olea europaea |
Whole oil |
4–200 |
Tyrosols, lignans (pinoresinol), phenolic acids, hydrolyzable tannins |
[20] |
|
Potato |
Solanum tuberosum |
Peel |
180–5000 |
Phenolic acids (chlorogenic acid) |
|
|
Flesh |
1–1000 |
||||
|
Total |
10–50 |
||||
|
Plum |
Prunus domestica |
Total |
130–240 |
Phenolic acids (chlorogenic acid), procyanidins, anthocyanins |
[20] |
|
Pomegranate |
Punica granatum |
Juice |
240 x |
Punicalagin (and ellagitannin) |
[22] |
|
Grapes, Red wine |
Vitis vinifera |
Final product |
25–300 x |
Phenolic acids, anthocyanins, tannins, stilbenes (resveratrol) |
[20] |
|
Wheat |
Triticum aestivum |
Whole grain |
85–220 |
Phenolic acids (hydroxybenzoic acids, hydroxycinnamic acids) |
[20] |
|
Spinach |
Spinacia oleracea |
Leaf |
30–290 |
Flavonols |
[20] |
Abbreviations: EC = epicatechin; EGCG = epigallocatechin gallate. x = In juices, wine, and other beverages: mg/100 mL. y = Concentration in mg/cm2. Note that the polyphenol content in purple potatoes is approximately five times higher than that in other varieties.
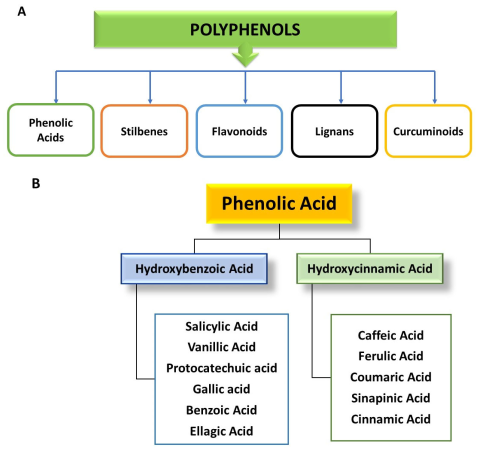
3. Classification of Polyphenols
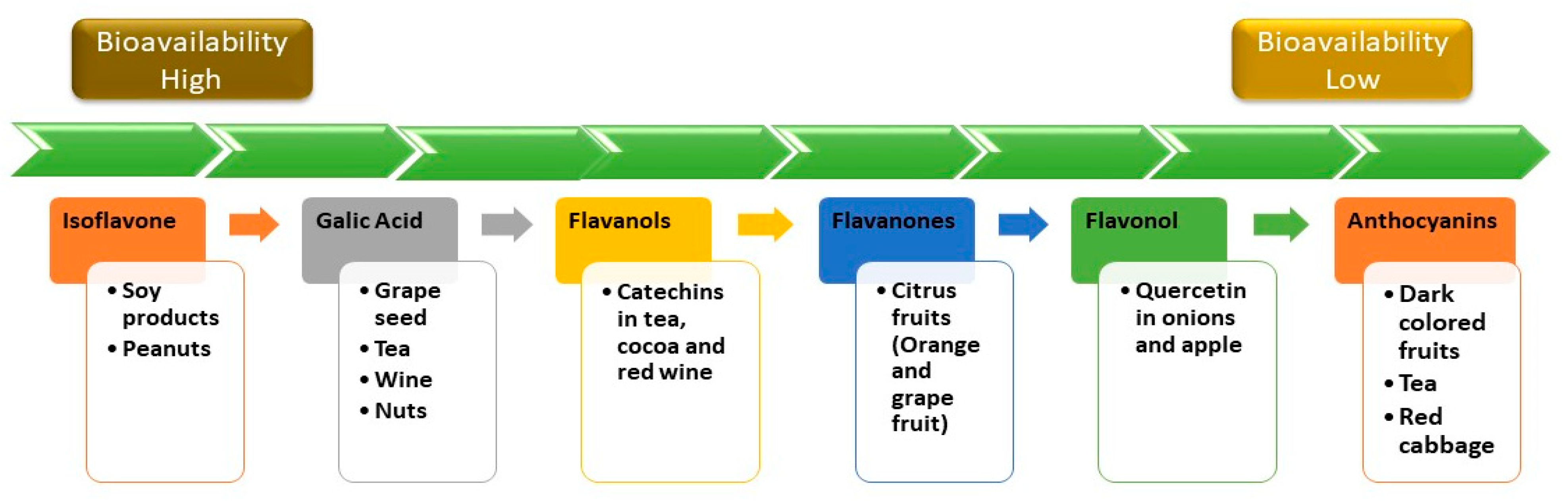
4. Bioavailability of Polyphenols
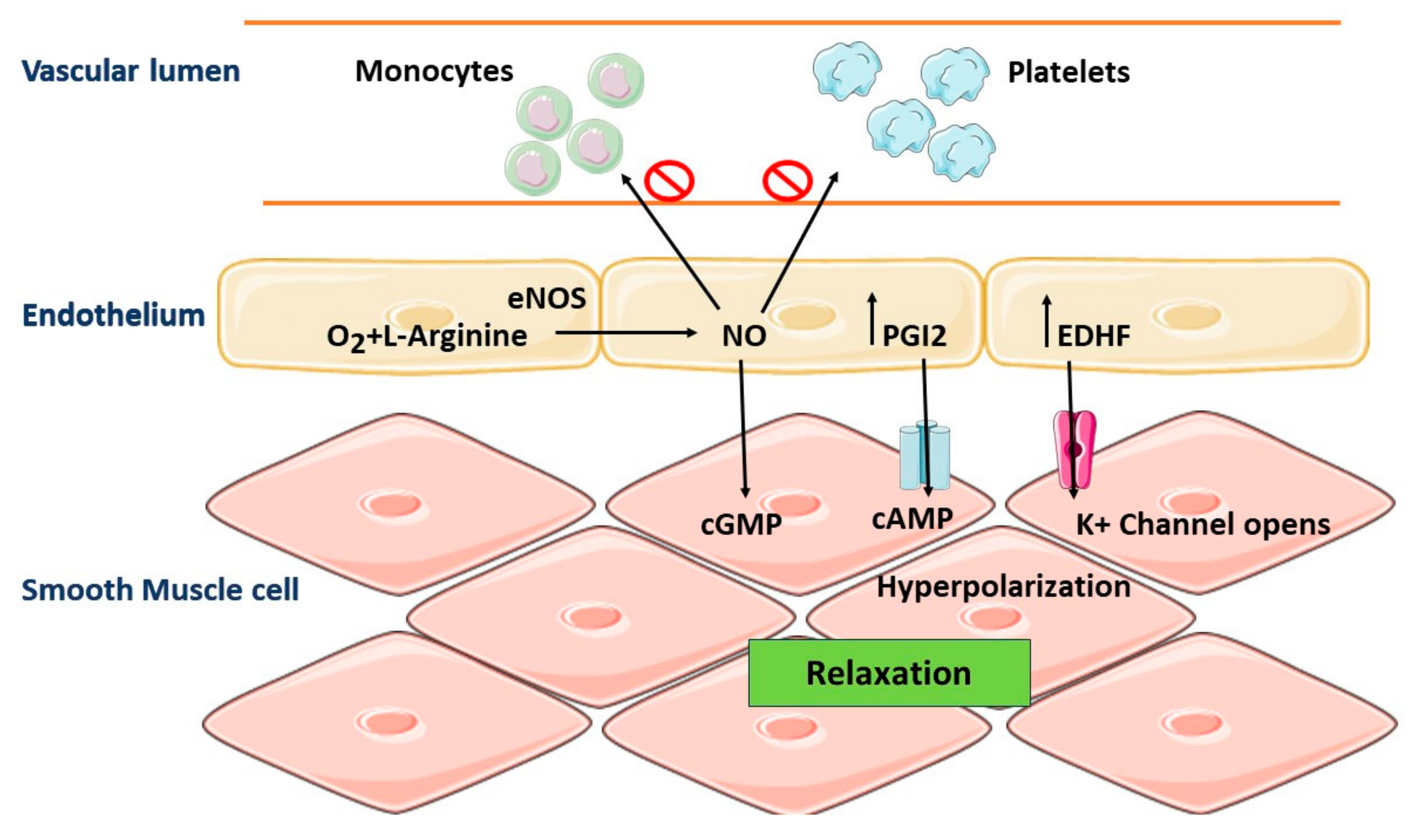
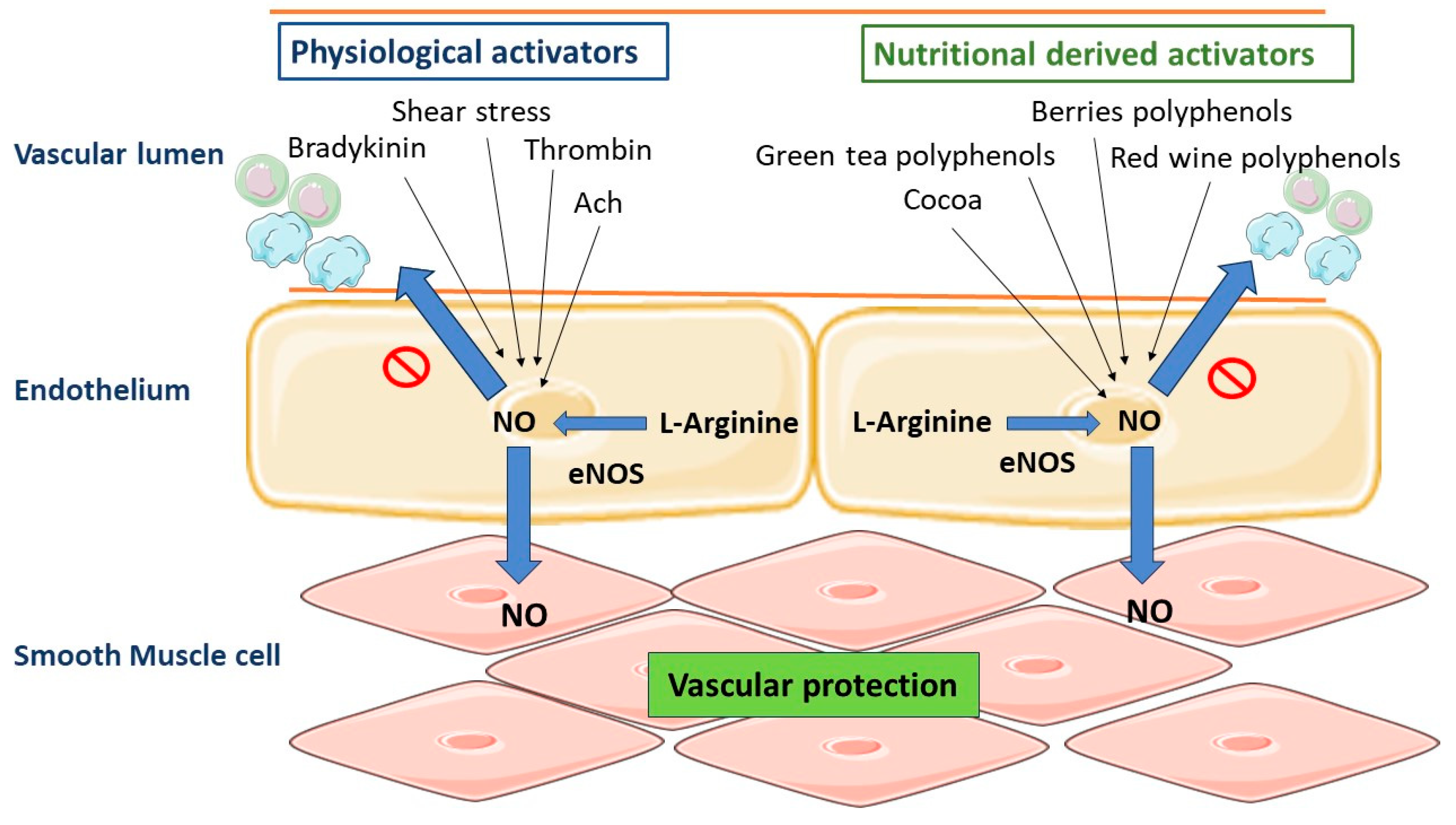
5. Role of Vascular Endothelium in the Regulation of Vascular Homeostasis
6. Pathophysiology: Oxidative Stress and CVD
7. Beneficial Effects of Polyphenols on Cardiovascular Disorders
7.1. Polyphenols as Antioxidant Therapy
7.2. Polyphenols and Vascular Tone
7.3. Polyphenols and Atherosclerosis
7.4. Polyphenols and Anti-Platelet Action
7.5. Polyphenols as Anti-Inflammatory Agents
References
- Goszcz, K.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Bioactive polyphenols and cardiovascular disease: Chemical antagonists, pharmacological agents or xenobiotics that drive an adaptive response? Br. J. Pharmacol. 2017, 174, 1209–1225.
- Alam, M.A. Anti-hypertensive effect of cereal antioxidant ferulic acid and its mechanism of action. Front. Nutr. 2019, 6, 121.
- Jamee Shahwan, A.; Abed, Y.; Desormais, I.; Magne, J.; Preux, P.M.; Aboyans, V.; Lacroix, P. Epidemiology of coronary artery disease and stroke and associated risk factors in Gaza community-Palestine. PLoS ONE 2019, 14, e0211131.
- Li, J.; Liao, R.; Zhang, S.; Weng, H.; Liu, Y.; Tao, T.; Yu, F.; Li, G.; Wu, J. Promising remedies for cardiovascular disease: Natural polyphenol ellagic acid and its metabolite urolithins. Phytomedicine 2023, 18, 154867.
- Sharifi-Rad, J.; Rodrigues, C.F.; Sharopov, F.; Docea, A.O.; Can Karaca, A.; Sharifi-Rad, M.; Kahveci Karıncaoglu, D.; Gülseren, G.; Şenol, E.; Demircan, E.; et al. Diet, lifestyle and cardiovascular diseases: Linking pathophysiology to cardioprotective effects of natural bioactive compounds. Int. J. Environ. Res. Public Health 2020, 17, 2326.
- Blauwet, L.A.; Cooper, L.T. Myocarditis. Prog. Cardiovasc. Dis. 2010, 52, 274–288.
- Wirtz, P.H.; von Känel, R. Psychological stress, inflammation, and coronary heart disease. Curr. Cardiol. Rep. 2017, 19, 111.
- Khan, J.; Deb, P.K.; Priya, S.; Medina, K.D.; Devi, R.; Walode, S.G.; Rudrapal, M. Dietary flavonoids: Cardioprotective potential with antioxidant effects and their pharmacokinetic, toxicological and therapeutic concerns. Molecules 2021, 26, 4021.
- Curry, S.J.; Krist, A.H.; Owens, D.K.; Barry, M.J.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W., Jr.; Kemper, A.R.; Kubik, M.; et al. Risk assessment for cardiovascular disease with nontraditional risk factors: US preventive services task force recommendation statement. JAMA 2018, 320, 272–280.
- Holvoet, P. Stress in obesity and associated metabolic and cardiovascular disorders. Scientifica 2012, 2012, 205027.
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341.
- Knowles, J.W.; Ashley, E.A. Cardiovascular disease: The rise of the genetic risk score. PLoS Med. 2018, 15, e1002546.
- Murray, M.; Dordevic, A.L.; Ryan, L.; Bonham, M.P. An emerging trend in functional foods for the prevention of cardiovascular disease and diabetes: Marine algal polyphenols. Crit. Rev. Food Sci. Nutr. 2018, 58, 1342–1358.
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747.
- Golovinskaia, O.; Wang, C.K. The hypoglycemic potential of phenolics from functional foods and their mechanisms. Food Sci. Hum. Wellness 2023, 12, 986–1007.
- Leuci, R.; Brunetti, L.; Poliseno, V.; Laghezza, A.; Loiodice, F.; Tortorella, P.; Piemontese, L. Natural compounds for the prevention and treatment of cardiovascular and neurodegenerative diseases. Foods 2021, 10, 29.
- Quiñones, M.; Miguel, M.; Aleixandre, A. Beneficial effects of polyphenols on cardiovascular disease. Pharmacol. Res. 2013, 68, 125–131.
- Wahid, M.; Saqib, F.; Chicea, L.; Ahmedah, H.T.; Sajer, B.H.; Marc, R.A.; Pop, O.L.; Moga, M.; Gavris, C. Metabolomics analysis delineates the therapeutic effects of hydroethanolic extract of Cucumis sativus L. seeds on hypertension and isoproterenol-induced myocardial infarction. Biomed. Pharmacother. 2022, 148, 112704.
- Bouayed, J.; Deußer, H.; Hoffmann, L.; Bohn, T. Bioaccessible and dialysable polyphenols in selected apple varieties following in vitro digestion vs. their native patterns. Food Chem. 2012, 131, 1466–1472.
- Rothwell, J.A.; Perez-Jimenez, J.; Neveu, V.; Medina-Remon, A.; M’hiri, N.; García-Lobato, P.; Manach, C.; Knox, C.; Eisner, R.; Wishart, D.S. Phenol-Explorer 3.0: A major update of the Phenol-Explorer database to incorporate data on the effects of food processing on polyphenol content. Database 2013, 2013, bat070.
- Deußer, H.; Guignard, C.; Hoffmann, L.; Evers, D. Polyphenol and glycoalkaloid contents in potato cultivars grown in Luxembourg. Food Chem. 2012, 135, 2814–2824.
- Rojanathammanee, L.; Puig, K.L.; Combs, C.K. Pomegranate polyphenols and extract inhibit nuclear factor of activated T-cell activity and microglial activation in vitro and in a transgenic mouse model of Alzheimer disease. J. Nutr. 2013, 143, 597–605.
- Rasouli, H.; Farzaei, M.H.; Khodarahmi, R. Polyphenols and their benefits: A review. Int. J. Food Prop. 2017, 20, 1700–1741.
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures; Oxford University Press: Oxford, UK, 2019; Volume 102, pp. 1397–1400.
- Prabhu, S.; Molath, A.; Choksi, H.; Kumar, S.; Mehra, R. Classifications of polyphenols and their potential application in human health and diseases. Int. J. Physiol, Nutr. Phys. Educ. 2021, 6, 293–301.
- Karas, D.; Ulrichová, J.; Valentová, K. Galloylation of polyphenols alters their biological activity. Food Chem. Toxicol. 2017, 105, 223–240.
- Bohn, T. Dietary factors affecting polyphenol bioavailability. Nutr. Rev. 2014, 72, 429–452.
- Gonzalez, S.; Fernandez, M.; Cuervo, A.; Lasheras, C. Dietary intake of polyphenols and major food sources in an institutionalised elderly population. J. Hum. Nutr. Diet. 2014, 27, 176–183.
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81 (Suppl. S1), 230s–242s.
- Crozier, A.; Del Rio, D.; Clifford, M.N. Bioavailability of dietary flavonoids and phenolic compounds. Mol. Aspects Med. 2010, 31, 446–467.
- Behl, T.; Bungau, S.; Kumar, K.; Zengin, G.; Khan, F.; Kumar, A.; Kaur, R.; Venkatachalam, T.; Tit, D.M.; Vesa, C.M.; et al. Pleotropic effects of polyphenols in cardiovascular system. Biomed. Pharmacother. 2020, 130, 110714.
- Laurent, C.; Besançon, P.; Caporiccio, B. Flavonoids from a grape seed extract interact with digestive secretions and intestinal cells as assessed in an in vitro digestion/Caco-2 cell culture model. Food Chem. 2007, 100, 1704–1712.
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561.
- Saqib, F.; Wahid, M.; AL-Huqail, A.A.; Ahmedah, H.T.; Bigiu, N.; Irimie, M.; Moga, M.; Marc, R.A.; Pop, O.L.; Chicea, L.M. Metabolomics based mechanistic insights to vasorelaxant and cardioprotective effect of ethanolic extract of Citrullus lanatus (Thunb.) Matsum. & Nakai. seeds in isoproterenol-induced myocardial infarction. Phytomedicine 2022, 100, 154069.
- Akimoto, S.; Mitsumata, M.; Sasaguri, T.; Yoshida, Y. Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21(Sdi1/Cip1/Waf1). Circ. Res. 2000, 86, 185–190.
- Sanches-Silva, A.; Testai, L.; Nabavi, S.F.; Battino, M.; Devi, K.P.; Tejada, S.; Sureda, A.; Xu, S.; Yousefi, B.; Majidinia, M.; et al. Therapeutic potential of polyphenols in cardiovascular diseases: Regulation of mTOR signaling pathway. Pharmacol. Res. 2020, 152, 104626.
- Ziche, M.; Morbidelli, L.; Masini, E.; Amerini, S.; Granger, H.J.; Maggi, C.A.; Geppetti, P.; Ledda, F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J. Clin. Investig. 1994, 94, 2036–2044.
- Matz, R.L.; Andriantsitohaina, R. Age-related endothelial dysfunction: Potential implications for pharmacotherapy. Drugs Aging 2003, 20, 527–550.
- Sofi, F.; Cesari, F.; Abbate, R.; Gensini, G.F.; Casini, A. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 9, 337.
- Rana, A.; Samtiya, M.; Dhewa, T.; Mishra, V.; Aluko, R.E. Health benefits of polyphenols: A concise review. J. Food Biochem. 2022, 46, e14264.
- Habauzit, V.; Morand, C. Evidence for a protective effect of polyphenols-containing foods on cardiovascular health: An update for clinicians. Ther. Adv. Chronic Dis. 2012, 3, 87–106.
- Khurana, S.; Venkataraman, K.; Hollingsworth, A.; Piche, M.; Tai, T.C. Polyphenols: Benefits to the cardiovascular system in health and in aging. Nutrients 2013, 5, 3779–3827.
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659.
- Bouayed, J.; Rammal, H.; Soulimani, R. Oxidative stress and anxiety: Relationship and cellular pathways. Oxid. Med. Cell. Longevity 2009, 2, 63–67.
- Lodovici, M.; Bigagli, E. Oxidative stress and air pollution exposure. J. Toxicol. 2011, 2011, 487074.
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124.
- Chan, S.M.; Cerni, C.; Passey, S.; Seow, H.J.; Bernardo, I.; van der Poel, C.; Dobric, A.; Brassington, K.; Selemidis, S.; Bozinovski, S.; et al. Cigarette smoking exacerbates skeletal muscle injury without compromising its regenerative capacity. Am. J. Respir. Cell Mol. Biol. 2020, 62, 217–230.
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247.
- Selvaraju, V.; Joshi, M.; Suresh, S.; Sanchez, J.A.; Maulik, N.; Maulik, G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease--an overview. Toxicol. Mech. Methods 2012, 22, 330–335.
- Le Brocq, M.; Leslie, S.J.; Milliken, P.; Megson, I.L. Endothelial dysfunction: From molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1631–1674.
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47 (Suppl. S8), C7–C12.
- Megson, I.L.; Whitfield, P.D.; Zabetakis, I. Lipids and cardiovascular disease: Where does dietary intervention sit alongside statin therapy? Food Funct. 2016, 7, 2603–2614.
- Loke, W.M.; Proudfoot, J.M.; Hodgson, J.M.; McKinley, A.J.; Hime, N.; Magat, M.; Stocker, R.; Croft, K.D. Specific dietary polyphenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler.; Thromb. Vasc. Biol. 2010, 30, 749–757.
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673.
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radical Biol. Med. 2011, 51, 978–992.
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255.
- Park, W.H.; Kim, S.H. Involvement of reactive oxygen species and glutathione in gallic acid-induced human umbilical vein endothelial cell death. Oncol. Rep. 2012, 28, 695–700.
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542.
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844.
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081.
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. S2), S170–S180.
- Curtin, J.F.; Donovan, M.; Cotter, T.G. Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 2002, 265, 49–72.
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovas. Res. 2015, 107, 321–330.
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519.
- Martínez-Cayuela, M. Oxygen free radicals and human disease. Biochimie 1995, 77, 147–161.
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53.
- Jennings, L.K. Mechanisms of platelet activation: Need for new strategies to protect against platelet-mediated atherothrombosis. Thromb. Haemost. 2009, 102, 248–257.
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949.
- Falk, E.; Shah, P.K.; Fuster, V. Coronary plaque disruption. Circulation 1995, 92, 657–671.
- Fasolo, F.; Di Gregoli, K.; Maegdefessel, L.; Johnson, J.L. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc. Res. 2019, 115, 1732–1756.
- Olas, B.; Wachowicz, B.; Tomczak, A.; Erler, J.; Stochmal, A.; Oleszek, W. Comparative anti-platelet and antioxidant properties of polyphenol-rich extracts from: Berries of Aronia melanocarpa, seeds of grape and bark of Yucca schidigera in vitro. Platelets 2008, 19, 70–77.
- Jagroop, I.A.; Kakafika, A.I.; Mikhailidis, D.P. Platelets and vascular risk: An option for treatment. Curr. Pharm. Des. 2007, 13, 1669–1683.
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511.
- Davì, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494.
- Jagroop, I.A.; Clatworthy, I.; Lewin, J.; Mikhailidis, D.P. Shape change in human platelets: Measurement with a channelyzer and visualisation by electron microscopy. Platelets 2000, 11, 28–32.
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435.
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Dukhyil, A.B.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary polyphenols and their role in oxidative stress-induced human diseases: Insights into protective effects, antioxidant potentials and mechanism (s) of action. Front. Pharmacol. 2022, 13, 283.
- Sies, H. Polyphenols and health: Update and perspectives. Arch. Biochem. Biophys. 2010, 501, 2–5.
- Hanif, S.; Shamim, U.; Ullah, M.F.; Azmi, A.S.; Bhat, S.H.; Hadi, S.M. The anthocyanidin delphinidin mobilizes endogenous copper ions from human lymphocytes leading to oxidative degradation of cellular DNA. Toxicology 2008, 249, 19–25.
- Puzserova, A.; Bernatova, I. Blood pressure regulation in stress: Focus on nitric oxide-dependent mechanisms. Physiol. Res. 2016, 65 (Suppl. S3), S309–S342.
- Andriambeloson, E.; Kleschyov, A.L.; Muller, B.; Beretz, A.; Stoclet, J.C.; Andriantsitohaina, R. Nitric oxide production and endothelium-dependent vasorelaxation induced by wine polyphenols in rat aorta. Br. J. Pharmacol. 1997, 120, 1053–1058.
- Duarte, J.; Andriambeloson, E.; Diebolt, M.; Andriantsitohaina, R. Wine polyphenols stimulate superoxide anion production to promote calcium signaling and endothelial-dependent vasodilatation. Physiol. Res. 2004, 53, 595–602.
- Zenebe, W.; Pechánová, O.; Andriantsitohaina, R. Red wine polyphenols induce vasorelaxation by increased nitric oxide bioactivity. Physiol. Res. 2003, 52, 425–432.
- Matsuo, S.; Nakamura, Y.; Takahashi, M.; Ouchi, Y.; Hosoda, K.; Nozawa, M.; Kinoshita, M. Effect of red wine and ethanol on production of nitric oxide in healthy subjects. Am. J. Cardiol. 2001, 87, 1029–1031.
- Ferrara, L.A.; Raimondi, A.S.; d’Episcopo, L.; Guida, L.; Dello Russo, A.; Marotta, T. Olive oil and reduced need for antihypertensive medications. Arch. Intern. Med. 2000, 160, 837–842.
- Diebolt, M.; Bucher, B.; Andriantsitohaina, R. Wine polyphenols decrease blood pressure, improve NO vasodilatation, and induce gene expression. Hypertension 2001, 38, 159–165.
- Andriambeloson, E.; Stoclet, J.C.; Andriantsitohaina, R. Mechanism of endothelial nitric oxide-dependent vasorelaxation induced by wine polyphenols in rat thoracic aorta. J. Cardiovasc. Pharmacol. 1999, 33, 248–254.
- Li, H.F.; Chen, S.A.; Wu, S.N. Evidence for the stimulatory effect of resveratrol on Ca2+-activated K+ current in vascular endothelial cells. Cardiovasc. Res. 2000, 45, 1035–1045.
- McKenna, E.; Smith, J.S.; Coll, K.E.; Mazack, E.K.; Mayer, E.J.; Antanavage, J.; Wiedmann, R.T.; Johnson, R.G., Jr. Dissociation of phospholamban regulation of cardiac sarcoplasmic reticulum Ca2+ ATPase by quercetin. J. Biol. Chem. 1996, 271, 24517–24525.
- Martin, S.; Andriambeloson, E.; Takeda, K.; Andriantsitohaina, R. Red wine polyphenols increase calcium in bovine aortic endothelial cells: A basis to elucidate signalling pathways leading to nitric oxide production. Br. J. Pharmacol. 2002, 135, 1579–1587.
- Ndiaye, M.; Chataigneau, M.; Lobysheva, I.; Chataigneau, T.; Schini-Kerth, V.B. Red wine polyphenol-induced, endothelium-dependent NO-mediated relaxation is due to the redox-sensitive PI3-kinase/Akt-dependent phosphorylation of endothelial NO-synthase in the isolated porcine coronary artery. FASEB J. 2005, 19, 455–457.
- Schramm, D.D.; Wang, J.F.; Holt, R.R.; Ensunsa, J.L.; Gonsalves, J.L.; Lazarus, S.A.; Schmitz, H.H.; German, J.B.; Keen, C.L. Chocolate procyanidins decrease the leukotriene-prostacyclin ratio in humans and human aortic endothelial cells. Am. J. Clin. Nutr. 2001, 73, 36–40.
- Fu, W.; Conklin, B.S.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Red wine prevents homocysteine-induced endothelial dysfunction in porcine coronary arteries. J. Surg. Res. 2003, 115, 82–91.
- Beretz, A.; Anton, R.; Cazenave, J.P. The effects of flavonoids on cyclic nucleotide phosphodiesterases. Prog. Clin. Biol. Res. 1986, 213, 281–296.
- Lugnier, C.; Schini, V.B. Characterization of cyclic nucleotide phosphodiesterases from cultured bovine aortic endothelial cells. Biochem. Pharmacol. 1990, 39, 75–84.
- Pechánová, O.; Bernátová, I.; Babál, P.; Martínez, M.C.; Kyselá, S.; Stvrtina, S.; Andriantsitohaina, R. Red wine polyphenols prevent cardiovascular alterations in L-NAME-induced hypertension. J. Hypertens. 2004, 22, 1551–1559.
- Aviram, M.; Rosenblat, M. Macrophage-mediated oxidation of extracellular low density lipoprotein requires an initial binding of the lipoprotein to its receptor. J. Lipid Res. 1994, 35, 385–398.
- Fuhrman, B.; Aviram, M. Flavonoids protect LDL from oxidation and attenuate atherosclerosis. Curr. Opin. Lipidol. 2001, 12, 41–48.
- Banach, M.; Markuszewski, L.; Zasłonka, J.; Grzegorczyk, J.; Okoński, P.; Jegier, B. The role of inflammation in the pathogenesis of atherosclerosis. Przegl. Epidemiol. 2004, 58, 663–670.
- Cordova, A.C.; Jackson, L.S.; Berke-Schlessel, D.W.; Sumpio, B.E. The cardiovascular protective effect of red wine. J. Am. Coll. Surg. 2005, 200, 428–439.
- El Haouari, M.; Rosado, J.A. Platelet signalling abnormalities in patients with type 2 diabetes mellitus: A review. Blood Cells Mol. Dis. 2008, 41, 119–123.
- Faggio, C.; Sureda, A.; Morabito, S.; Sanches-Silva, A.; Mocan, A.; Nabavi, S.F.; Nabavi, S.M. Flavonoids and platelet aggregation: A brief review. Eur. J. Pharmacol. 2017, 807, 91–101.
- Hubbard, G.P.; Stevens, J.M.; Cicmil, M.; Sage, T.; Jordan, P.A.; Williams, C.M.; Lovegrove, J.A.; Gibbins, J.M. Quercetin inhibits collagen-stimulated platelet activation through inhibition of multiple components of the glycoprotein VI signaling pathway. J. Thromb. Haemost. 2003, 1, 1079–1088.
- Vanhoutte, P.M. Endothelial dysfunction: The first step toward coronary arteriosclerosis. Circ. J. 2009, 73, 595–601.
- de Gaetano, G.; De Curtis, A.; di Castelnuovo, A.; Donati, M.B.; Iacoviello, L.; Rotondo, S. Antithrombotic effect of polyphenols in experimental models: A mechanism of reduced vascular risk by moderate wine consumption. Ann. N. Y. Acad. Sci. 2002, 957, 174–188.
- Mladěnka, P.; Zatloukalová, L.; Filipský, T.; Hrdina, R. Cardiovascular effects of flavonoids are not caused only by direct antioxidant activity. Free Radic. Biol. Med. 2010, 49, 963–975.
- Demrow, H.S.; Slane, P.R.; Folts, J.D. Administration of wine and grape juice inhibits in vivo platelet activity and thrombosis in stenosed canine coronary arteries. Circulation 1995, 91, 1182–1188.
- Hamid, A.A.; Aminuddin, A.; Yunus, M.H.M.; Murthy, J.K.; Hui, C.K.; Ugusman, A. Antioxidative and anti-inflammatory activities of Polygonum minus: A review of literature. Rev. Cardiovasc. Med. 2020, 21, 275–287.
- Choy, K.W.; Murugan, D.; Leong, X.F.; Abas, R.; Alias, A.; Mustafa, M.R. Flavonoids as natural anti-inflammatory agents targeting nuclear factor-kappa B (NFκB) signaling in cardiovascular diseases: A mini-review. Front. Pharmacol. 2019, 10, 1295.
- Dias, M.C.; Pinto, D.C.; Silva, A.M. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377.
- Liao, H.; Ye, J.; Gao, L.; Liu, Y. The main bioactive compounds of Scutellaria baicalensis Georgi. for alleviation of inflammatory cytokines: A comprehensive review. Biomed. Pharmacother. 2021, 133, 110917.
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as potential anti-inflammatory molecules: A review. Molecules 2022, 27, 2901.
- Krauth, V.; Bruno, F.; Pace, S.; Jordan, P.M.; Temml, V.; Romano, M.P.; Khan, H.; Schuster, D.; Rossi, A.; Filosa, R.; et al. Highly potent and selective 5-lipoxygenase inhibition by new, simple heteroaryl-substituted catechols for treatment of inflammation. Biochem. Pharmacol. 2023, 208, 115385.
- Sychrová, A.; Škovranová, G.; Čulenová, M.; Bittner Fialová, S. Prenylated flavonoids in topical infections and wound healing. Molecules 2022, 27, 4491.
- Martinez, J.; Moreno, J.J. Effect of resveratrol, a natural polyphenolic compound, on reactive oxygen species and prostaglandin production. Biochem. Pharmacol. 2000, 59, 865–870.
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H quinone oxidoreductase (NQO1): An enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019, 39, BSR20180459.

