 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vicente Javier Clemente-Suárez | -- | 8505 | 2023-09-14 10:17:36 | | | |
| 2 | Lindsay Dong | Meta information modification | 8505 | 2023-09-15 03:17:10 | | | | |
| 3 | Lindsay Dong | Meta information modification | 8505 | 2023-09-19 04:22:38 | | | | |
| 4 | Lindsay Dong | + 1 word(s) | 8506 | 2023-10-24 02:06:11 | | |
Video Upload Options
Mitochondria play a vital role in maintaining cellular energy homeostasis, regulating apoptosis, and controlling redox signaling. Dysfunction of mitochondria has been implicated in the pathogenesis of various brain diseases, including neurodegenerative disorders, stroke, and psychiatric illnesses.
1. Introduction
2. Molecular Mechanisms in the Development of Diseases
2.1. Mitochondria Bioenergetics
2.2. Calcium Signalling
2.3. Cell Death
3. Neurodegenerative Disorders and Mitochondrial Dysfunction
3.1. Mitochondrial Dysfunctional Functioning in Neurodegenerative Disorders
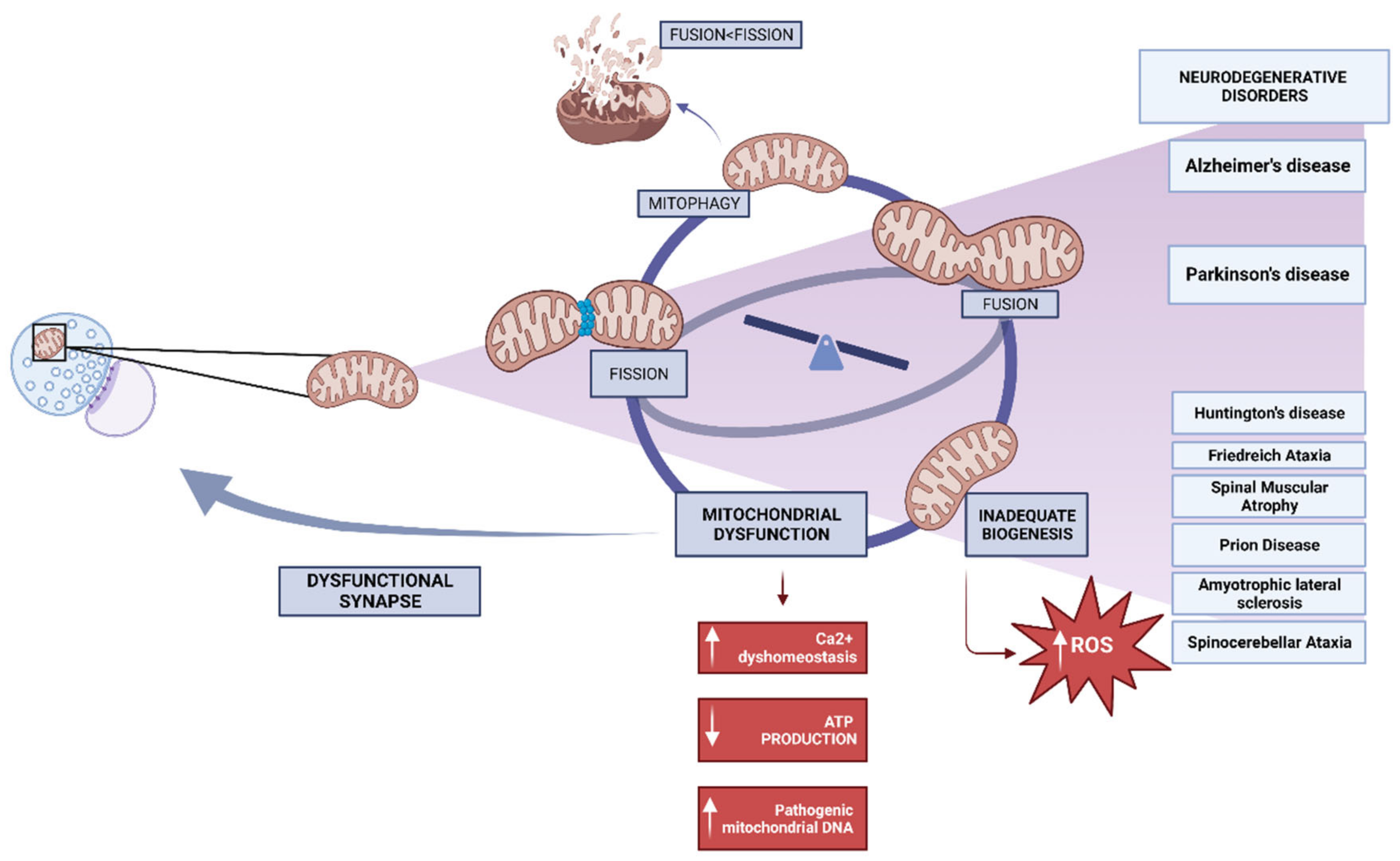
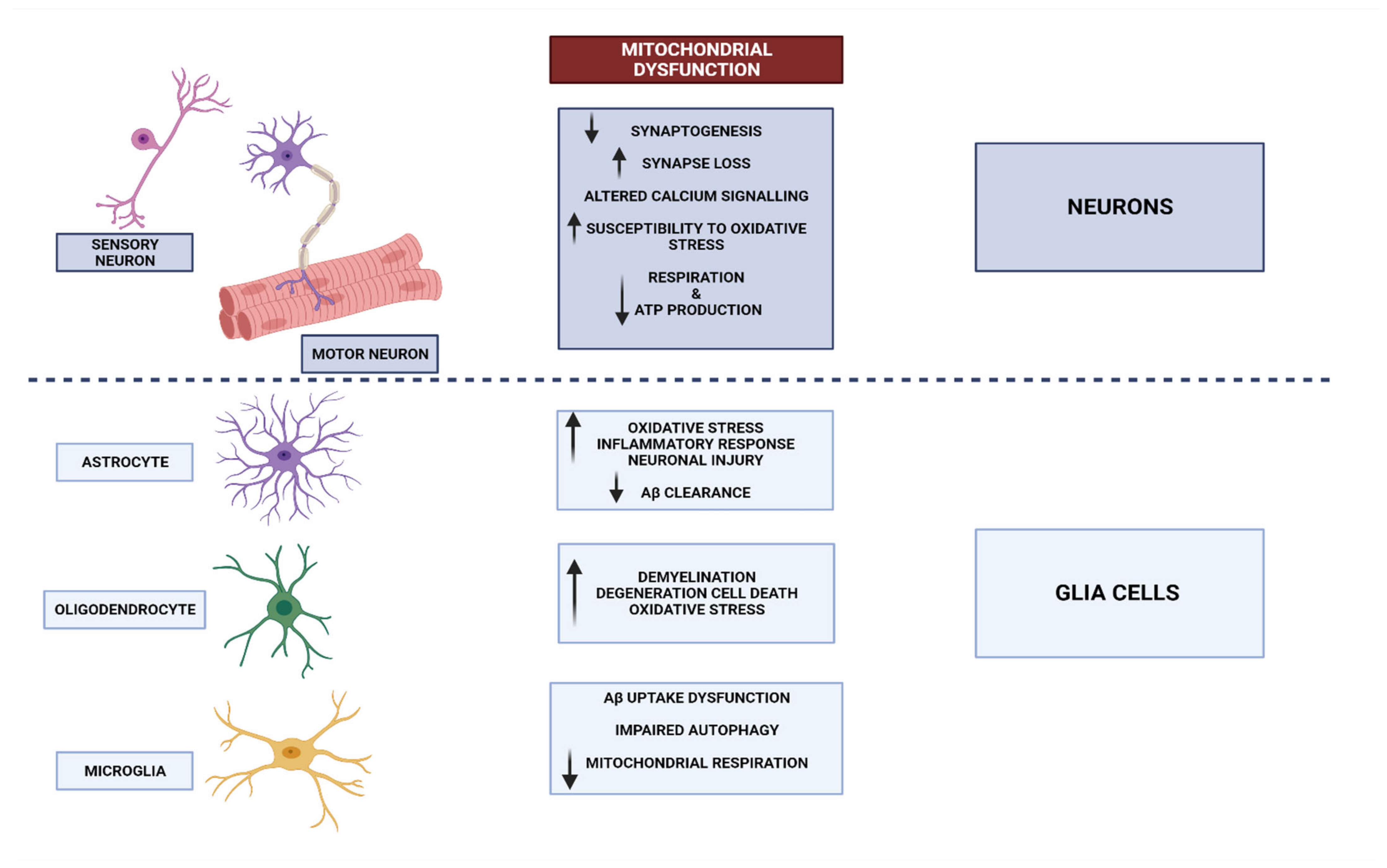
3.1.1. Mitochondrial Dysfunction in Alzheimer’s Disease
3.1.2. Mitochondrial Dysfunction in Parkinson’s Disease
3.1.3. Mitochondrial Dysfunction in Other Neurodegenerative Disease
4. Mitochondrial DNA Mutations in Brain Disease
5. Impaired Oxidative Phosphorylation and Brain Disease
6. Mitochondrial Dynamics in Neuronal Health
7. Calcium Dysregulation and Its Impact on Mitochondria
Cellular dysfunctions affecting structures such as mitochondria and the endoplasmic reticulum, along with increased oxidative stress and dysregulation of calcium homeostasis, are linked to the pathogenesis of Alzheimer’s disease (AD). The sodium-calcium exchanger (NCLX) is crucial for cellular metabolism regulation in both the plasma membrane and mitochondria. The dependency between energy metabolism and intracellular calcium levels is proposed as one of the first modifiable deteriorations in brain ageing. Therefore, it is essential to determine whether modifying NCLX activity to increase cellular metabolism and mitochondrial calcium content could prevent neuronal impairment and death [32].
8. Reactive Oxygen Species and Mitochondrial Dysfunction
9. Inflammation and Mitochondrial Impairment in Brain Disease
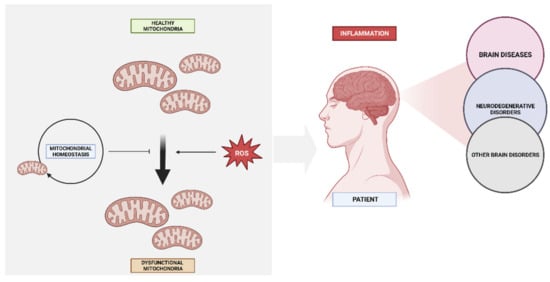
10. Mitochondria and Neurotransmitter Systems in Psychiatric Illnesses
11. Mitochondrial Biomarkers in Brain Disease
11.1. Mitochondrial Biomarkers in Neurodegenerative Disorders
11.2. Mitochondrial Biomarkers in Psychiatric Illnesses
12. Therapeutic Strategies: Mitochondrial Protective Agents
13. Metabolic Modulators and Mitochondrial Function in Brain Diseases
14. Gene Therapy for Restoring Mitochondrial Function
References
- San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782.
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723.
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Tornero-Aguilera, J.F.; Ruisoto, P.; Mielgo-Ayuso, J. Inflammation in COVID-19 and the Effects of Non-Pharmacological Interventions during the Pandemic: A Review. Int. J. Mol. Sci. 2022, 23, 15584.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080.
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044.
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261.
- Barros, M.H.; McStay, G.P. Modular biogenesis of mitochondrial respiratory complexes. Mitochondrion 2019, 50, 94–114.
- Zhou, Q.; Zhai, Y.; Lou, J.; Liu, M.; Pang, X.; Sun, F. Thiabendazole inhibits ubiquinone reduction activity of mitochondrial respiratory complex II via a water molecule mediated binding feature. Protein Cell 2011, 2, 531–542.
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514.
- Belhadj Slimen, I.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523.
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and Beta-Amyloid. Different Targets, Same Players: Calcium, Free Radicals and Mitochondria in the Mechanism of Neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115.
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6.
- Juárez, D.; González-Ravé, J.M.; Rubio-Arias, J.Á.; Clemente Suárez, V.J.; Valencia, M.; Abian Vicente, J. Isokinetic leg strength and power in elite handball players. J. Hum. Kinet. 2014, 41, 227–233.
- Bruni, F. Mitochondria: From Physiology to Pathology. Life 2021, 11, 991.
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4.
- Puthalakath, H.; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002, 9, 505–512.
- Pellegrini, L.; Passer, B.J.; Tabaton, M.; Ganjei, J.K.; D’Adamio, L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999, 274, 21011–21016.
- van den Heuvel, M.P.; Sporns, O. Network hubs in the human brain. Trends Cogn. Sci. 2013, 17, 683–696.
- Pino, A.; Fumagalli, G.; Bifari, F.; Decimo, I. New neurons in adult brain: Distribution, molecular mechanisms and therapies. Biochem. Pharmacol. 2017, 141, 4–22.
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84.
- Kaminsky, N.; Bihari, O.; Kanner, S.; Barzilai, A. Connecting Malfunctioning Glial Cells and Brain Degenera-tive Disorders. Genom. Proteom. Bioinform. 2016, 14, 155–165.
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211.
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia Reprogram Metabolic Profiles for Phenotype and Function Changes in Central Nervous System. Neurobiol. Dis. 2021, 152, 105290.
- Ganat, Y.M.; Silbereis, J.; Cave, C.; Ngu, H.; Anderson, G.M.; Ohkubo, Y.; Ment, L.R.; Vaccarino, F.M. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J. Neurosci. 2006, 26, 8609–8621.
- Fakhoury, M. Immune-mediated processes in neurodegeneration: Where do we stand? J. Neurol. 2016, 263, 1683–1701.
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735.
- Martin, J.B. Molecular Basis of the Neurodegenerative Disorders. N. Engl. J. Med. 1999, 340, 1970–1980.
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Ap-proaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851.
- Clemente-Suárez, V.J.; Ramos-Campo, D.J.; Mielgo-Ayuso, J.; Dalamitros, A.A.; Nikolaidis, P.A.; Hormeño-Holgado, A.; Tornero-Aguilera, J.F. Nutrition in the Actual COVID-19 Pandemic. A Narrative Review. Nutrients 2021, 13, 1924.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coac-tivator PGC-1. Cell 1999, 98, 115–124.
- Chew, K.; Zhao, L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes 2021, 12, 1246.
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313.
- Chen, S.-D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ische-mia. Int. J. Mol. Sci. 2011, 12, 7199–7215.
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029.
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53.
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115.
- Kim, S.; Pajarillo, E.; Nyarko-Danquah, I.; Aschner, M.; Lee, E. Role of Astrocytes in Parkinson’s Disease Asso-ciated with Genetic Mutations and Neurotoxicants. Cells 2023, 12, 622.
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508.
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835.
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549.
- Pohland, M.; Pellowska, M.; Asseburg, H.; Hagl, S.; Reutzel, M.; Joppe, A.; Berressem, D.; Eckert, S.H.; Wurglics, M.; Schubert-Zsilavecz, M.; et al. MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 18.
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767.
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; De Waal, R.M.W.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328.
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochon-drially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372.
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32.
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2017, 25, 24–34.
- MacDonald, M.E.; Barnes, G.; Srinidhi, J.; Duyao, M.P.; Ambrose, C.M.; Myers, R.H.; Gray, J.; Conneally, P.M.; Young, A.; Penney, J. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Genet. 1993, 30, 982–986.
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362.
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46.
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra97.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268.
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680.
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106.
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188.
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784.
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240.
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25.
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392.
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980.
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen. Med. 2020, 5, 22.
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237.
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639.
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660.
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267.
- Hamblet, N.S.; Castora, F.J. Elevated levels of the Kearns–Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutat. Res. 1997, 379, 253–262.
- Sequeira, A.; Martin, M.V.; Rollins, B.; Moon, E.A.; Bunney, W.E.; Macciardi, F.; Lupoli, S.; Smith, E.N.; Kelsoe, J.; Magnan, C.N.; et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet. 2012, 3, 103.
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37.
- Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Hung, P.-L.; Hsu, M.-H.; Wang, T.-J.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Chang, P.-C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65.
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608.
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322.
- Garrido-Pérez, N.; Vela-Sebastián, A.; López-Gallardo, E.; Emperador, S.; Iglesias, E.; Meade, P.; Jiménez-Mallebrera, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome. Int. J. Mol. Sci. 2020, 21, 3374.
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242.
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491.
- Yuan, X.; Liu, Y.; Bijonowski, B.M.; Tsai, A.-C.; Fu, Q.; Logan, T.M.; Ma, T.; Li, Y. NAD(+)/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun. Biol. 2020, 3, 774.
- Clemente-Suárez, V.J.; Robles-Pérez, J.J. Psycho-physiological response of soldiers in urban combat. An. Psicol. 2013, 29, 598–603.
- Van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 93.
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090.
- Xie, H.-R.; Hu, L.-S.; Li, G.-Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092.
- Agholme, L.; Lindström, T.; Kågedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 2010, 20, 1069–1082.
- Devine, M.J.; Birsa, N.; Kittler, J.T. Miro sculpts mitochondrial dynamics in neuronal health and disease. Neurobiol. Dis. 2016, 90, 27–34.
- Avdoshina, V.; Fields, J.A.; Castellano, P.; Dedoni, S.; Palchik, G.; Trejo, M.; Adame, A.; Rockenstein, E.; Eugenin, E.; Masliah, E.; et al. The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotox. Res. 2016, 29, 583–593.
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55.
- Crews, L.; Tsigelny, I.; Hashimoto, M.; Masliah, E. Role of synucleins in Alzheimer’s disease. Neurotoxicity Res. 2009, 16, 306–317.
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430.
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal 2011, 14, 459–468.
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postep. Hig. Med. Dosw. Online 2016, 70, 1150–1165.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303.
- Clemente-Suárez, V.J.; Mielgo-Ayuso, J.; Quiles, J.L.; Varela-Lopez, A.; Aranda, P. Effect of α-tocopherol megadoses on hematologic parameters and antioxidant capacity of rats in an ultraendurance probe. Physiol. Int. 2017, 104, 291–300.
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298.
- García-Giménez, J.L.; Pallardó, F.V. Maintenance of glutathione levels and its importance in epigenetic regulation. Front. Pharmacol. 2014, 5, 88.
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2015.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal 2020, 32, 677–700.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52.
- Tõugu, V.; Palumaa, P. Coordination of zinc ions to the key proteins of neurodegenerative diseases: Aβ, APP, α-synuclein and PrP. Coord. Chem. Rev. 2012, 256, 2219–2224.
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206.
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128.
- Wang, P.; Wang, Z.-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290.
- Tiiman, A.; Palumaa, P.; Tõugu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 95–121.
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942.
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643.
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874.
- Wang, Y.; Pan, Y.; Li, H. What is brain health and why is it important? BMJ 2020, 371, m3683.
- Currais, A. Ageing and inflammation—A central role for mitochondria in brain health and disease. Ageing Res. Rev. 2015, 21, 30–42.
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937.
- Yan, F.; Tang, H.; Wang, L.; Huang, L.; Zhang, J. Editorial: Mitochondrial Dysfunction in Stroke. Front. Aging Neurosci. 2022, 14, 888952.
- Widmann, C.N.; Heneka, M.T. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014, 13, 630–636.
- Zsurka, G.; Kunz, W.S. Mitochondrial involvement in neurodegenerative diseases. IUBMB Life 2013, 65, 263–272.
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2020, 217, 136–147.
- Spiers, J.G.; Chen, H.-J.C.; Bourgognon, J.-M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free. Radic. Biol. Med. 2019, 134, 468–483.
- Foyet, S.H.; Balmus, I.-M.; Hervé, N.A.H.; Emmanuel, A.A.; Guenne, S.; Kiendrebéogo, M.; Ciobica, A. Ethnopharmacological approaches in mood and anxiety disorders. The relevance of the oxidative stress status. J. Complement. Integr. Med. 2017, 14.
- Vaváková, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid. Med. Cell. Longev. 2015, 2015, 898393.
- Madireddy, S.; Madireddy, S. Therapeutic Interventions to Mitigate Mitochondrial Dysfunction and Oxidative Stress-Induced Damage in Patients with Bipolar Disorder. Int. J. Mol. Sci. 2022, 23, 1844.
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 8881770.
- Kato, T.A.; Yamauchi, Y.; Horikawa, H.; Monji, A.; Mizoguchi, Y.; Seki, Y.; Hayakawa, K.; Utsumi, H.; Kanba, K. Neurotransmitters, psychotropic drugs and microglia: Clinical implications for psychiatry. Curr. Med. Chem. 2013, 20, 331–344.
- Ye, N.; Song, Z.; Zhang, A. Dual ligands targeting dopamine D2 and serotonin 5-HT1A receptors as new antipsychotical or anti-Parkinsonian agents. Curr. Med. Chem. 2013, 21, 437–457.
- Clemente-Suárez, V.J. Multidisciplinary intervention in the treatment of mixed anxiety and depression disorder. Physiol. Behav. 2020, 219, 112858.
- Lopresti, A.L.; Hood, S.D.; Drummond, P.D. A review of lifestyle factors that contribute to important pathways associated with major depression: Diet, sleep and exercise. J. Affect. Disord. 2013, 148, 12–27.
- Sacher, J.; Rekkas, P.V.; Wilson, A.A.; Houle, S.; Romano, L.; Hamidi, J.; Rusjan, P.; Fan, I.; Stewart, D.E.; Meyer, J.H. Relationship of monoamine oxidase—A distribution volume to postpartum depression and postpartum crying. Neuropsychopharmacology 2015, 40, 429–435.
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Mielgo-Ayuso, J.; Martínez-Guardado, I.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Antioxidants and Sports Performance. Nutrients 2023, 15, 2371.
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069.
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21.
- Bemeur, C. Oxidative Stress in the Central Nervous System Complications of Chronic Liver Failure. In Studies on Hepatic Disorders; Springer: Berlin/Heidelberg, Germany, 2015; pp. 357–370.
- Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. Int. J. Mol. Sci. 2023, 24, 7221.
- Mandal, P.K.; Gaur, S.; Roy, R.G.; Samkaria, A.; Ingole, R.; Goel, A. Schizophrenia, Bipolar and Major Depressive Disorders: Overview of Clinical Features, Neurotransmitter Alterations, Pharmacological Interventions, and Impact of Oxidative Stress in the Disease Process. ACS Chem. Neurosci. 2022, 13, 2784–2802.
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68.
- Khadimallah, I.; Jenni, R.; Cabungcal, J.-H.; Cleusix, M.; Fournier, M.; Beard, E.; Klauser, P.; Knebel, J.-F.; Murray, M.M.; Retsa, C.; et al. Mitochondrial, exosomal miR137-COX6A2 and gamma synchrony as biomarkers of parvalbumin interneurons, psychopathology, and neurocognition in schizophrenia. Mol. Psychiatry 2022, 27, 1192–1204.
- Carreira Míguez, M.; Clemente Suárez, V.J. Physical activity levels affect mental health and behavior in men. J. Men’s Health 2023, 1, 12.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127.
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178.
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8.
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Yang, X.; Dai, G.; Li, G.; Yang, E.S. Coenzyme Q10 reduces β-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010, 41, 110–113.
- Fouad, G.I. Combination of Omega 3 and Coenzyme Q10 Exerts Neuroprotective Potential Against Hypercholesterolemia-Induced Alzheimer’s-Like Disease in Rats. Neurochem. Res. 2020, 45, 1142–1155.
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 147, 14–21.
- Bagheri, S.; Haddadi, R.; Saki, S.; Kourosh-Arami, M.; Rashno, M.; Mojaver, A.; Komaki, A. Neuroprotective effects of coenzyme Q10 on neurological diseases: A review article. Front. Neurosci. 2023, 17, 1188839.
- Jardim, F.R.; De Rossi, F.T.; Nascimento, M.X.; Barros, R.G.d.S.; Borges, P.A.; Prescilio, I.C.; De Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101.
- Alikiaii, B.; Bagherniya, M.; Askari, G.; Sathyapalan, T.; Sahebkar, A. Evaluation of the effect of curcumin on pneumonia: A systematic review of preclinical studies. Phytotherapy Res. 2021, 35, 1939–1952.
- Bagherniya, M.; Nobili, V.; Blesso, C.N.; Sahebkar, A. Medicinal plants and bioactive natural compounds in the treatment of non-alcoholic fatty liver disease: A clinical review. Pharmacol. Res. 2018, 130, 213–240.
- Ghandadi, M.; Sahebkar, A. Curcumin: An Effective Inhibitor of Interleukin-6. Curr. Pharm. Des. 2017, 23, 921–931.
- Mollazadeh, H.; Cicero, A.F.G.; Blesso, C.N.; Pirro, M.; Majeed, M.; Sahebkar, A. Immune modulation by curcumin: The role of interleukin-10. Crit. Rev. Food Sci. Nutr. 2019, 59, 89–101.
- Panahi, Y.; Ahmadi, Y.; Teymouri, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a potential candidate for treating hyperlipidemia: A review of cellular and metabolic mechanisms. J. Cell. Physiol. 2018, 233, 141–152.
- Teymouri, M.; Pirro, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a multifaceted compound against human papilloma virus infection and cervical cancers: A review of chemistry, cellular, molecular, and preclinical features. Biofactors 2017, 43, 331–346.
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717.
- Plioplys, A.V.; Plioplys, S. Serum levels of carnitine in chronic fatigue syndrome: Clinical correlates. Neuropsychobiology 1995, 32, 132–138.
- Latham, L.E.; Wang, C.; Patterson, T.A.; Slikker, W.; Liu, F. Neuroprotective Effects of Carnitine and Its Potential Application to Ameliorate Neurotoxicity. Chem. Res. Toxicol. 2021, 34, 1208–1222.
- Vidal-Casariego, A.; Burgos-Peláez, R.; Martínez-Faedo, C.; Calvo-Gracia, F.; Valero-Zanuy, M.; Luengo-Pérez, L.M.; Cuerda-Compés, C. Metabolic Effects of L-carnitine on Type 2 Diabetes Mellitus: Systematic Review and Meta-analysis. Exp. Clin. Endocrinol. Diabetes 2013, 121, 234–238.
- Förster, L.; Indra, D.; Rosenberger, K.; Zver, L.; Hofbauer, R. L-carnitine exerts a nutrigenomic effect via direct modulation of nuclear receptor signaling in adipocytes, hepatocytes and SKMC, demonstrating its nutritional impact. Nutr. Res. 2021, 85, 84–98.
- Virmani, M.; Caso, V.; Spadoni, A.; Rossi, S.; Russo, F.; Gaetani, F. The action of acetyl-L-carnitine on the neurotoxicity evoked by amyloid fragments and peroxide on primary rat cortical neurones. Ann. N. Y. Acad. Sci. 2001, 939, 162–178.
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 398–408.
- Dhitavat, S.; Ortiz, D.; Shea, T.B.; Rivera, E.R. Acetyl-L-carnitine protects against amyloid-beta neurotoxicity: Roles of oxidative buffering and ATP levels. Neurochem. Res. 2002, 27, 501–505.
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 8848.
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343.
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560.
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810.
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091.
- Arduino, D.M.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism modulation: A new therapeutic approach for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 105–119.
- Hagen, T.M.; Ingersoll, R.T.; Lykkesfeldt, J.; Liu, J.; Wehr, C.M.; Vinarsky, V.; Bartholomew, J.C.; Ames, B.N. (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 1999, 13, 411–418.
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959.
- Tai, S.; Zheng, Q.; Zhai, S.; Cai, T.; Xu, L.; Yang, L.; Jiao, L.; Zhang, C. Alpha-lipoic acid mediates clearance of iron accumulation by regulating iron metabolism in a Parkinson’s disease model induced by 6-OHDA. Front. Neurosci. 2020, 14, 612.
- Liu, L.; Yang, S.; Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP(+)-induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 2021, 45, 422–431.
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 38, 111–120.
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural Transm. Suppl. 2007, 72, 189–193.
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. J. Neurodegener. Dis. 2013, 2013, 1–7.
- Smith, R.A.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352.
- Mounsey, R.B.; Teismann, P. Mitochondrial dysfunction in Parkinson’s disease: Pathogenesis and neuroprotection. Park. Dis. 2011, 2011, 617472.
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649.
- Silva-Pinheiro, P.; Minczuk, M. The potential of mitochondrial genome engineering. Nat. Rev. Genet. 2022, 23, 199–214.
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586.
- Zhu, Y. Advances in CRISPR/Cas9. BioMed Res. Int. 2022, 2022, 9978571.

