Mitochondria play a vital role in maintaining cellular energy homeostasis, regulating apoptosis, and controlling redox signaling. Dysfunction of mitochondria has been implicated in the pathogenesis of various brain diseases, including neurodegenerative disorders, stroke, and psychiatric illnesses.
- mitochondria
- brain disease
- neurodegenerative disorders
- mitochondrial DNA mutations
- oxidative phosphorylation
- mitochondrial dynamics
1. Introduction
2. Molecular Mechanisms in the Development of Diseases
Mitochondria are essential for normal cellular function because they are responsible for energy production in eukaryotes, as well as phospholipid and heme synthesis, calcium homeostasis, apoptotic activation, and cell demise [1][3]. As a comprehensive overview of multiple pathological processes, the following will analyse and identify the molecular processes occurring in the mitochondria that are involved in specific common patterns in the pathological process’s development. Mitochondrial diseases are a group of genetic disorders characterised by defects in oxidative phosphorylation and caused by mutations in genes that encode structural mitochondrial proteins or proteins involved in mitochondrial function in nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). Mitochondrial diseases are the most prevalent category of inherited metabolic disorders and one of the most prevalent types of inherited neurological diseases [4][5]. During normal neuronal development, programmed cell death (PCD) signalling events occur in a spatially and temporally restricted manner to establish the neural architecture and shape the CNS. In the pathogenesis of numerous neurological diseases, abnormalities in PCD signalling cascades, such as apoptosis, necroptosis, pyroptosis, ferroptosis, cell death associated with autophagy, and unprogrammed necrosis, can be observed. These cell fatalities can be triggered in response to intracellular or extracellular stimuli and inflammatory processes. A prevalent characteristic of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease is the aberrant activation of PCD pathways, which leads to the unwarranted loss of neuronal cells and function [6].2.1. Mitochondria Bioenergetics
The mitochondria contain the main enzymatic systems required to complete the oxidation of sugars, lipids, and proteins to produce adenosine triphosphate (ATP), which can be used for energy production. Each of these three substrates can be catabolized to acetyl-CoA, which then enters the citric acid cycle in the mitochondrial matrix, the first of these processes [1]. Following glycolysis in the cytosol, glucose enters the mitochondria as pyruvate. Pyruvate dehydrogenase aids in the conversion of pyruvate to acetyl-CoA. Inside the mitochondria, beta oxidation converts fatty acids to acetyl-CoA, while various enzymes convert specific amino acids into pyruvate, acetyl-CoA, or directly into specific citric acid cycle intermediates [7]. In the citric acid cycle, also known as the tricarboxylic acid (TCA) or Krebs cycle, the acetyl group of acetyl-CoA is transferred to oxaloacetate to form the six carbon molecule citrate. In seven successive enzymatic steps, citrate is oxidised back to oxaloacetate, with the excess carbon removed as two molecules of carbon dioxide and the electrons transferred to cofactors nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). The oxaloacetate is now free to rejoin the cycle, while NADH and FADH2 transport the liberated energy to the mitochondrial electron transport chain [8]. The electron transport chain, also known as the respiratory chain, is comprised of a series of multisubunit protein complexes embedded in the interior mitochondrial membrane [9]. The electrons removed from the citric acid cycle by NADH and FADH2 are used to enable the pumping of protons from the matrix to the intermembrane space, thereby generating a potential difference across the inner mitochondrial membrane [10]. By binding to the largest of the respiratory complexes, NADH dehydrogenase, or complex I, NADH provides free energy to the electron transport chain. NADH transfers a pair of electrons, which are transported away from the citric acid cycle, to a flavin mononucleotide prosthetic group located inside the hydrophilic arm of complex I. Subsequently, the electrons are sent up the arm through a sequence of iron-sulphur clusters until they reach the lipid-soluble redox carrier known as coenzyme Q [3]. The transfer of four protons across the inner mitochondrial membrane is associated with the transport of electrons from NADH via the complex. NADH must diffuse into complex I in order to furnish the electrons it delivers to electron transport. Conversely, succinate dehydrogenase, the enzyme responsible for catalysing the reduction of FAD to FADH2 in the citric acid cycle, is an integral component of the electron transport chain. Commonly referred to as complex II, this enzyme with a molecular weight of 123 kDa is situated on the inner mitochondrial membrane, similar to complex I [11]. The reduced form of coenzyme Q may traverse the inner mitochondrial membrane without hindrance and afterwards transfer its electrons to cytochrome c reductase, the third complex of the electron transport chain. This reduction can occur via either complex I or complex II. The final outcome of the electrons transferred through the electron transport chain is the conversion of oxygen into water. This phenomenon takes place in the fourth complex, known as cytochrome c oxidase [12]. During the progression of electrons along the electron transport chain, there is a gradual reduction in their free energy, which coincides with a consistent rise in the redox potential of the carriers involved. Ultimately, this process culminates with oxygen, possessing the highest redox potential among all the carriers. The energy that is liberated as the electron moves down the free energy “staircase” serves as the driving force for the thermodynamically unfavourable process of proton pumping against their concentration gradient at complexes I, III, and IV [3]. After the completion of the citric acid cycle and the electron transport chain, the final step in converting the energy stored in substrate molecules into ATP, which serves as the universal “energy currency”, involves the linkage of the membrane voltage of approximately 200 mV to the phosphorylation of adenosine diphosphate (ADP). In addition to causing the decoupling of membrane voltage from ATP generation, proton leakage is also accompanied by the leakage of electrons from the complexes of the electron transport chain [9]. The premature release of electrons enables their direct transfer to oxygen, bypassing the conventional pathway of electron transfer to oxygen for water formation at complex IV and therefore generating superoxide. Superoxide has a high level of reactivity, rendering it significantly detrimental to cellular function by inducing what is commonly referred to as “oxidative stress” [13]. The involvement of oxidative stress has been suggested in several pathological conditions, ranging from atherosclerosis and diabetes to neurodegenerative disorders and cancer.2.2. Calcium Signalling
Mitochondria are in constant communication with the cytosol in order to coordinate the equilibrium between the energy needs of the cell and the energy produced by oxidative phosphorylation [3]. This is orchestrated predominantly by calcium signalling between the cytosol and matrix. Ca2+ signalling is essential for the majority of cellular ‘activation states’: Ca2+ signals regulate the majority of processes that are associated with increased energy demands—secretion, contraction, motility, and electrical excitability—all of which necessitate an increase in energy provision and are typically accompanied by an increase in cytosolic Ca2+ concentration ([Ca2+]c) [14][15]. In the inner mitochondrial membrane, mitochondria exhibit a Ca2+ uptake pathway, the mitochondrial calcium uniporter (MCU), which is a Ca2+ selective channel. As Ca2+ moves down its electrochemical potential gradient into the matrix, a rise in local [Ca2+]c stimulates mitochondrial Ca2+ uptake [16]. The increase in matrix Ca2+ concentration ([Ca2+]m) stimulates the three rate-limiting enzymes of the TCA cycle: pyruvate, α-ketogluterate (also known as oxogluterate), and NAD-isocitrate dehydrogenases. A rise in [Ca2+]m also appears to upregulate ATP synthase, although the mechanism remains unknown. Together, these mechanisms increase the supply of NADH to the respiratory chain, respiration, and ultimately the rate of ATP synthesis. The increase in [Ca2+]c also activates the glutamate/aspartate transporter (ARALAR) on the inner mitochondrial membrane, thereby increasing substrate supply; this pathway does not require specific mitochondrial Ca2+ uptake and is therefore independent of mitochondrial bioenergetic competence [15]. Thus, these pathways function together to elegantly and simply match energy supply and demand. In the majority of cells, the efflux of Ca2+ from the mitochondrial matrix, driven by a Na+/Ca2+ exchanger, is relatively sluggish, such that the change in [Ca2+]m significantly outlasts the change in [Ca2+]c, and the metabolic response likely matches the time course of the change in [Ca2+]m.2.3. Cell Death
Significant evidence supports a role for cell death in the pathogenesis of numerous brain and peripheral nervous system diseases. However, it is still unclear whether defects in cell death signalling and neuronal cell death are a primary or secondary response to the attacks that cause these diseases, as well as how different PCD pathways and additional processes interact to cause the death of neuronal cells and other cell types in these diseases [6][17]. The regulation of the intrinsic route involves the modulation of pro- and anti-apoptotic members belonging to the BCL-2 protein family [18]. The preservation of cell viability in healthy cells is facilitated by a group of proteins known as anti-apoptotic proteins, namely BCL-2, BCL-XL, MCL-1, BCL-W, and A1/BFL1 [19]. These proteins fulfil this role by inhibiting the crucial mediators of cell death, BAX and BAK. The BH3-only proteins, including BIM, PUMA, BID, BMF, BAD, HRK, BIK, and NOXA, which play a crucial role in initiating apoptosis, are seen to undergo transcriptional or post-transcriptional upregulation in reaction to intracellular stress such as growth factor deprivation, DNA damage, and ER stress [19]. The BH3-only proteins have a strong binding affinity for anti-apoptotic BCL-2 proteins, resulting in the release of BAX and BAK. Several studies have demonstrated that some BH3-only proteins have the ability to directly activate BAX and BAK. Upon being activated, BAX and BAK proteins undergo oligomerization, leading to mitochondrial outer membrane permeabilization (MOMP). This process results in the release of cytochrome c and Smac/DIABLO from the mitochondria. The apoptogenic factors facilitate the initiation of the caspase cascade, which subsequently triggers the cleavage of many proteins, ultimately culminating in cellular implosion [6]. The available evidence supporting the involvement of apoptosis in neuronal cell death in AD is constrained. Nevertheless, according to the immuno-histochemical labelling of neurons, it has been postulated that intracellular Aβ has the potential to trigger apoptosis via the p53-dependent transcriptional upregulation of BAX [20]. Necroptosis is a kind of programmed cell death characterised by cellular lysis, which has the potential to induce an inflammatory response. Necroptosis may be triggered by the activation of TNFR1, TLRs, and specific additional receptors in instances when the function of caspase-8 is impeded by pharmaceutical substances or viral inhibitors. The mechanism under consideration entails the activation of receptor-interacting serine/threonine protein kinase 1 (RIPK1) by autophosphorylation [6].3. Neurodegenerative Disorders and Mitochondrial Dysfunction
The information transmitted by neurons is crucial to normal brain operation [21]. Neurons can be found in many other tissues and organs beyond the brain [22]. Glia, also known as neuroglia, constitute a significant proportion of cellular entities within the intricate framework of the nervous system [23]. While neurons exhibit excitability and play a crucial role in information processing, glial cells take on the complete responsibility for maintaining homeostasis and defending the central nervous system (CNS). Various types of glial cells have been implicated in both normal and pathological brain function [24]. Astrocytes elegantly extend their processes throughout grey matter, gracefully enveloping synapses with their presence. These remarkable cells play a pivotal role in maintaining the delicate balance of ions and neurotransmitters, ensuring a harmonious environment within the central nervous system. Furthermore, they diligently oversee the intricate process of synaptogenesis, orchestrating the formation of vital connections between neurons. It is worth noting that astrocytes also possess the remarkable ability to secrete various substances, thus earning them the esteemed title of secretory cells within the CNS [25]. Microglia are responsible for regulating the elimination of apoptotic neurons and play a role in influencing the formation of synaptic connections during brain development [26]. In pathological conditions, these microglia become activated and contribute to the occurrence of neuroinflammation and myelination, which play a crucial role in shaping the connectome. The essential glial functions, as well as the interaction between glial cells and neurons, delineate the functioning of the brain in both states of well-being and pathology [24] (Figure 1). Neural stem cells generate most neurons during development, but their numbers drastically fall once we reach adulthood [27]. Although neuronal death is inevitable, neurodegeneration remains a major health issue because of its role in the pathophysiology of certain neurological disorders [28]. Neurodegeneration is associated with the accumulation of proteins with physiochemical modifications, synapse dysfunction, and neural network malfunction [29]. Related to this, neurodegenerative disorders (NDs) are the medical terminologies used to describe diseases that exhibit the characteristics of neurodegeneration. Some of the most common NDs seen in clinical praxis are Parkinson’s disease, Alzheimer’s disease, prion disease, amyotrophic lateral sclerosis, motor neuron disease, spinal muscular atrophy, Huntington’s disease, and spinocerebellar ataxia [30][31].3.1. Mitochondrial Dysfunctional Functioning in Neurodegenerative Disorders
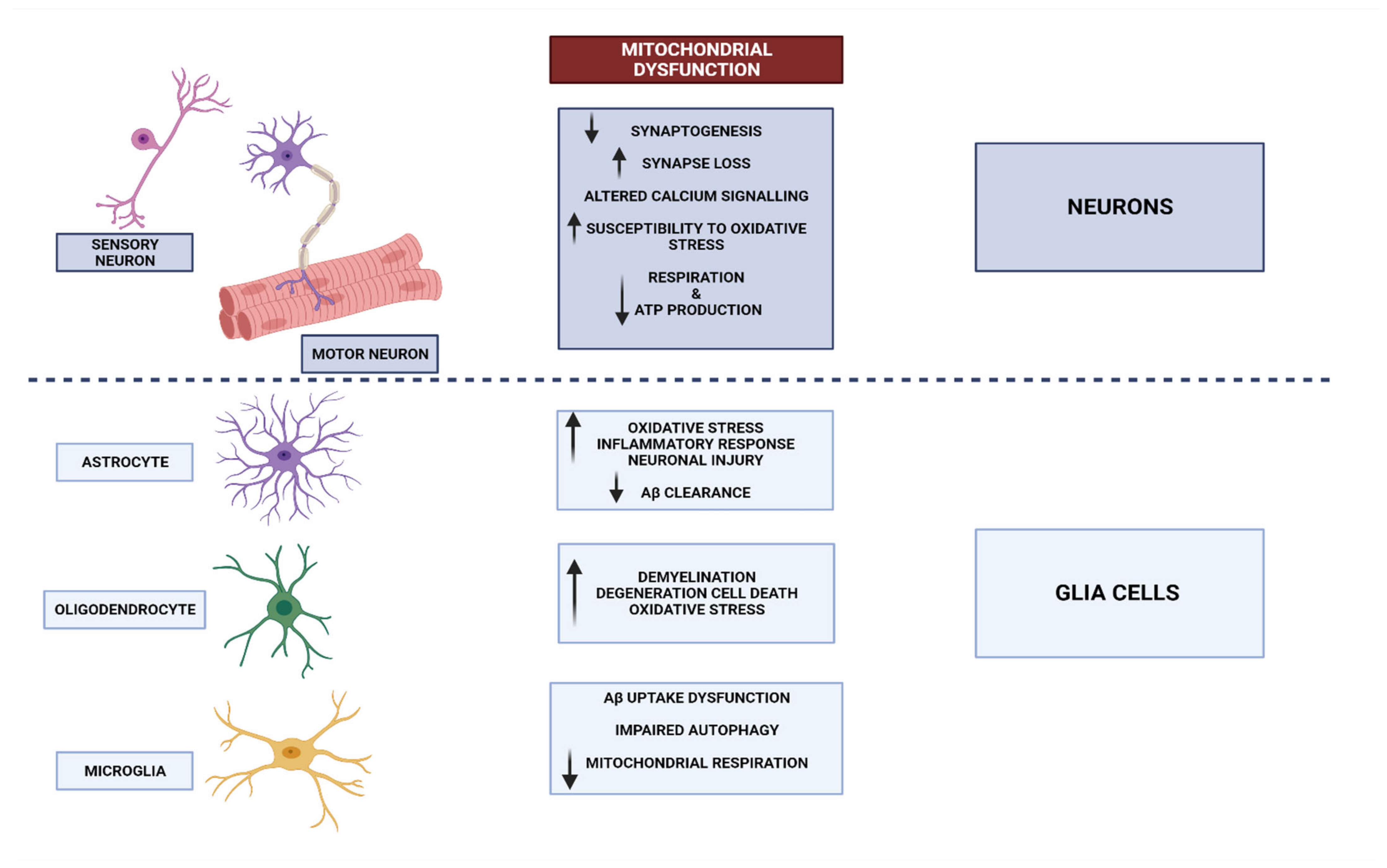
The phenomenon known as mitochondrial biogenesis refers to the intricate process wherein fresh mitochondria are engendered through the growth and division of pre-existing mitochondria. This intricate process ultimately leads to a notable augmentation in the mitochondrial mass within a given cell [32]. Protein synthesis from the mitochondrial and nuclear genomes, as well as replication of mitochondrial DNA (mtDNA), are all part of this process, which is regulated by peroxisome proliferator-activated receptor gamma (PPAR γ) and PGC1 α [33]. PGC1 α functions as a master regulator that integrates and coordinates the activity of nuclear respiratory factors 1 and 2 (NRF1-2) and mitochondrial transcription factor A (TFAM) [34]. By attaching to TFAM promotor sites, they set off the mitochondrial transcription and replication processes [34][35]. The reduction in ATP synthesis, compromise in mitochondrial integrity, and elevation in oxidative stress are all consequences observed when these processes become imbalanced [36]. Deficiencies in the process of mitochondrial biogenesis may potentially contribute to the manifestation of mitochondrial dysfunction in various neurodegenerative conditions, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Friedreich ataxia, among other notable examples [37]. The neurodegenerative process is believed to involve oxidative stress from ROS generation, but the downstream steps of mitochondrial malfunction leading to neuronal cell death are not well known. Mitochondria are both the main generators of ROS and the principal targets of increased exposure to them. Moreover, with the progression and increased accessibility of imaging technologies, the previously held notion of mitochondria as stationary, bean-shaped cellular structures has been invalidated. The mitochondria, being highly mobile and dynamic organelles, exhibit a remarkable ability to undergo fusion and fragmentation. This intricate process, referred to as “mitochondrial dynamics”, is tightly regulated and occurs in response to various environmental stimuli [38]. In a cell, mitochondria that have fused together can be seen as a tubular network with many connections. The efficient transfer of mtDNA during mitochondrial biogenesis and a metabolic rate that matches the cell’s energy needs are both made possible by this network. Consequently, the health of a cell depends on a finely tuned balance between fusion and fission (Figure 2). The focus of scientific inquiry pertaining to mitochondrial dysfunction and its implications in neurological disorders has predominantly revolved around neurons. On the contrary, the involvement of malfunctioning mitochondria in the functioning of glial cells and its potential impact on neuronal homeostasis and brain function have received limited attention in the scientific literature. However, based on the available evidence, it is evident that the maintenance of mitochondrial health within glial cells plays a crucial role in the proper functioning of neurons [39] (Figure 1).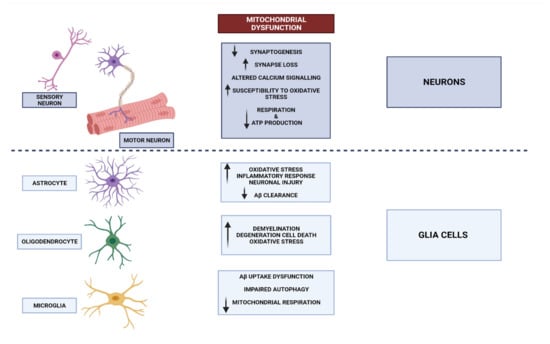
3.1.1. Mitochondrial Dysfunction in Alzheimer’s Disease
3.1.2. Mitochondrial Dysfunction in Parkinson’s Disease
Parkinson’s disease (PD), the second most prevalent neurodegenerative disorder, is characterised by symptoms including bradykinesia, resting tremor, postural instability, akinesia, and occasionally dementia. The disease is characterised by a range of mitochondrial abnormalities, including impairment of the electron transport chain, changes in mitochondrial morphology and dynamics, mutations in mitochondrial DNA, and disturbances in calcium homeostasis. These abnormalities are depicted in Figure 21 [48]. From a pathological standpoint, these characteristics are indicative of a more pronounced decline in dopaminergic (DA) neurons and the existence of Lewy bodies containing α -synuclein in the substantia nigra pars compacta (SNpc).3.1.3. Mitochondrial Dysfunction in Other Neurodegenerative Disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterised by involuntary movements, psychiatric disturbances, and subcortical dementia [49]. It is caused by an expansion of CAG repeats that confers a toxic function on the Huntingtin (Htt) protein [50]. Although HD’s neuropathology manifests itself in multiple brain regions, it is most obviously seen in the striatum, where it is defined by selective cell death. Although the processes are unknown, defects in mitochondrial biogenesis may contribute to the striatum’s susceptibility to Huntington’s disease. Transcriptional sup-pression by mHtt-dependent sequestration of transcription factors CREB/TAF4 has been found in the striatum of HD mouse models and human patients, leading to lower levels of PGC1 α mRNA and protein [51][52]. Oxidative stress, such as lipid peroxidation and DNA oxidative damage, is significantly elevated in HD, thereby amplifying the formation of amyloidogenic mutant huntingtin (mHtt) aggregates and possibly contributing to the vicious cycle in the pathophysiology of HD [53].4. Mitochondrial DNA Mutations in Brain Disease
The mitochondria have the characteristic that their cellular function is regulated by a double energy system. On the one hand, it has nuclear DNA that it shares with the rest of the cells of the organism, and on the other hand, it has DNA of its own, the mtDNA, which is composed of thirty-seven genes, thirteen peptides, two ribosomal RNAs, and twenty-two ribonucleic acids, or transfer RNAs [54]. It is precisely this unique origin of mtDNA from the egg that explains why mitochondrial pathologies present maternal patterns in the exclusive alterations of mtDNA [55]. It is known that the main function of mitochondria is to facilitate cellular respiration for metabolic energy, in addition to other functions involved in pathways such as cytogenesis, thermogenesis, intracellular Ca2 regulation, or fatty acid oxidation [56]. Although evidence has been found that there are mutations in genes encoded in nDNA, it has been established in recent decades that the main mutations in mtDNA lead to dysregulation and malfunction of the oxidative phosphorylation system [57]. All these mutations cause the so-called mitochondrial pathologies or mitochondrial encephalomyopathies, still considered “rare diseases” since their prevalence is low, i.e., less than 5/10,000 inhabitants [58][59]. However, the impact on the paediatric population is very serious, this being the most frequent age range of onset. Similarly, diagnosis in adults is becoming more common [60]. These diseases are still a challenge for medicine today and are characterised by very heterogeneous clinical manifestations with alterations in different tissues and organs. However, they share the alteration of oxidative phosphorylation with the presence of encephalopathy and lesions at the muscular level [61][62]. In relation to the brain pathologies that have been determined so far to be associated with mitochondrial mutations, scholars find symptomatology such as seizures, convulsions, and reduced intellectual capacity, although the symptomatology will depend on the phenotype, the mutation, and the level of complementation with normal mtDNA [63][64][65]. Mitochondriopathies present a wide variety of phenotypes, depending on their association with point mutations or when insertions, deletions, or both appear and the DNA is restructured. Leber’s hereditary optic neuropathy is included in this categorisation and is associated with bilateral optic atrophy and other symptoms such as dystonia. Early onset (12–30 years) and more prevalent in males than in females [66]. The most frequently identified mutations are A-G 11,778 in the ND4 gene and G-A 3460 in the ND1 gene [67][68]. In relation to diseases due to insertions or deletions, or both, that produce restructuring of mtDNA, the Kearns–Sayre syndromes, Pearson syndromes, as well as the Leukoencephalopathic syndromes and chronic sporadic progressive external ophthalmoplegia are related [69]. In this line, a recent study has been able to identify psychiatric and neurodegenerative pathologies and diseases associated with ageing related to genetic alterations in the mitochondrial DNA of the brain [70]. In AD and PD, the results indicated elevated levels of heteroplasmy as well as a higher presence of variants in the parietal cortex, cerebellum, and hippocampus in AD patients compared to the control group. Specifically in AD, the most frequent mutation is the 4977 bp deletion (m.8470_13477del) in the temporal lobe, which occurs with a higher mutation load in the caudate nucleus than in the frontal gyrus [71]. Understanding the pathological mechanisms of mitochondrial DNA mutations should be useful to design effective therapeutic interventions in each patient, knowing the heterogeneity of the clinical presentation [72]. Currently, there is no treatment for these pathologies, with some exceptions in which the mutation appears in a single organ and is amenable to transplantation [73].5. Impaired Oxidative Phosphorylation and Brain Disease
Cellular bioenergetics has studied cellular processes over the last few decades and has been able to confirm that most of these processes are induced by the hydrolysis of adenosine triphosphate (ATP). Through the prolonged oxidation of fatty acids, amino acids, and monosaccharides, ATP homeostasis is maintained [74]. The energy of these elements is transferred mainly as electrons to flavin adenine dinucleotide (FAD) or nicotinamide adenine dinucleotide (NAD+), although some are converted directly to ATP [56]. The OXPHOS system ensures higher energy utilisation thanks to its dual activity. On the one hand, the electron transport chain includes ATP synthase, coenzyme Q (CoQ10), cytochrome c (Cytc), and the respiratory complexes CI-IV, proteins that are in the inner membrane of the mitochondrion and that are originated by the unification of units encoded in both the mtDNA and the DNA of the mitochondrial nucleus [57]. On the other hand, the propagation of protons from the mitochondrial matrix to the intermembrane space occurs and facilitates the emergence of the proton-motive force, essential for ATP synthesis. The last process in this system involves the utilisation of electrochemical energy to propagate ATP synthesis [75][76]. Sirtuins are one of the primary metabolic sensors in the cell, and this is possible because they are more sensitive to metabolic state, increasing the NAD+/NADH (reduced nicotinamine adenine dinucleotide) ratio after physical activity or fasting, stimulating deacetylation by sirtuins [77][78]. Regarding the energy metabolism of the cells of the Central Nervous System (CNS), neurons need a large amount of ATP and are dependent on the constant circulation of glucose [26][79]. In addition, these cells have no glycogen reserves. Therefore, it is important to maintain the functionality of the mitochondria in these cells since a deterioration of this functionality is related to neurodegeneration, both in old age and in diseases such as AD [80]. When there is an impairment in oxidative phosphorylation mediated by the OXPHOS system, the antioxidant mechanisms of the biological system are imbalanced, and this will lead to alterations in metabolic pathways and their activity, as well as the accumulation of intracellular aggregates, apoptosis, excitotoxicity, and mitochondrial dysfunction [81]. In this line, recent studies based on animal models, specifically an in vitro model of the neuroblastoma cell line SH-SY5Y, have been able to find neuronal loss in the substantia nigra leading to dopaminergic hypofunction [82]. It is also interesting to know that many AD patients present parkinsonian symptomatology as well as significant damage to the cholinergic system. The presence of significant cortical cholinergic denervation suggests that these cells are more vulnerable to pathology [82][83].6. Mitochondrial Dynamics in Neuronal Health
Neurons are complex, excitable, and polarised cells that require enormous amounts of energy. This demand for adenosine triphosphate (ATP) is primarily met through oxidative phosphorylation in mitochondria. Mitochondria play an essential role in neuronal function, undergoing constant cycles of fission/fusion at sites of energy demand. The morphology of mitochondria varies between axonal and dendritic compartments. Axons, which are smaller due to their self-correcting capacity that prevents extremes in size, exhibit more dynamic behaviour than dendrites. Recent advances have improved our understanding of mitochondrial dynamics and neuronal function. In particular, the mitochondrial permeability transition (MPT) governs mitochondrial dynamics and its transformation. Balanced fission/fusion is crucial for transportation. The knowledge of the importance of mitochondria for neuronal health is increasingly consolidated. In fact, mitochondria are essential for maintaining local energy supplies and attenuating calcium flux in neurons. Devine et al. established in their 2016 review the dependence of miro proteins on coordinating mitochondrial trafficking, dynamics, and turnover in neuronal homeostasis [84]. Specifically, mitochondrial dynamics are crucial for adjusting mitochondrial activity and efficiency. Initially, mitochondria were considered isolated and immobile organelles, but they are now recognised as highly active and interconnected systems within cells. This movement not only affects mitochondrial structure but also impacts the activity of organelles present in the cytoskeleton. This process, specifically regulated by mitochondrial fission/fusion cycles, is known as mitochondrial dynamics. These dynamics play a prominent role in cellular homeostasis, and abnormalities in mitochondrial fission/fusion proteins are associated with diseases, particularly age-related diseases. Altered expression of certain dynamic mitochondrial proteins is linked to ageing and age-related disorders, connecting ageing with a decline in mitochondrial activity and defective mitochondrial biogenesis. In addition to ageing, various neurodegenerative disorders are determined by mitochondrial dysfunction. Avdoshina et al. [85] demonstrated that the gp120 protein of HIV reduces mitochondrial capacity and distribution. These alterations in mitochondrial distribution initiate neurodegeneration because the disruption of mitochondrial trafficking negatively affects energy distribution in synapses. It is noteworthy that these consequences occur even in the absence of the virus, suggesting that this protein alone is sufficient to initiate definitive neurodegenerative mechanisms, interacting with different endogenous neurotoxins or different pathophysiological insults [85]. Studying the characteristics of mitochondrial dynamics, including fission/fusion, transport, biogenesis, and degradation, is essential for understanding neuronal health. In fact, mitochondrial dynamics and bioenergetics are interconnected. This is particularly relevant for neurons, as they depend on mitochondria for bioenergetic support. Van Laar and Berman [86] highlight the importance of the interplay between bioenergetics and mitochondrial dynamics in neurodegenerative processes. Different forms and structural characteristics of mitochondria correlate with the diverse bioenergetic needs of the tissues they occupy. Neurons rely on mitochondrial oxidative phosphorylation as their primary energy supply. Preserving a functional population of healthy mitochondria is therefore particularly challenging at the neuronal level.7. Calcium Dysregulation and Its Impact on Mitochondria
Cellular dysfunctions affecting structures such as mitochondria and the endoplasmic reticulum, along with increased oxidative stress and dysregulation of calcium homeostasis, are linked to the pathogenesis of Alzheimer’s disease (AD). The sodium-calcium exchanger (NCLX) is crucial for cellular metabolism regulation in both the plasma membrane and mitochondria. The dependency between energy metabolism and intracellular calcium levels is proposed as one of the first modifiable deteriorations in brain ageing. Therefore, it is essential to determine whether modifying NCLX activity to increase cellular metabolism and mitochondrial calcium content could prevent neuronal impairment and death [32].
Both PD and AD are neurodegenerative diseases originating from misfolding and aggregation of key proteins. Oligomeric beta-amyloid and alpha-synuclein stimulate mitochondrial depolarization due to excessive calcium and free radical generation, leading to the opening of the mitochondrial permeability transition pore and ultimately cell death [87]. Among neurodegenerative diseases related to ageing, AD is the most prevalent and is associated with the deposition of beta-amyloid protein in senile plaques. Intracellular calcium acts as a second messenger and is essential for regulating neuronal functions, action potential, and synaptic plasticity. Abnormal aggregation of beta-amyloid peptide (Aβ) is proposed as a precursor to degenerative events in cholinergic neurons. Therefore, alterations in calcium homeostasis are investigated to understand the processes of Aβ-induced neurodegeneration. Neurofibrillary tangles (NFTs), aggregates of hyperphosphorylated tau protein, give rise to the “tau hypothesis”, where NFTs are considered the main culprits in neuronal loss and memory impairment due to axonal transport alterations in AD. Scientific evidence indicates that excitatory synaptic dysregulation participates in neurodegeneration. In fact, dysregulation of mitochondrial calcium is associated with post-synaptic excitatory neurodegeneration. It was documented how excitatory lesions associated with neurodegeneration in PD, AD, amyotrophic lateral sclerosis (ALS), and HD. Therefore, therapeutic research should also focus on preserving mitochondrial function through calcium homeostasis regulation [88]. In this context, the primary excitatory neurotransmitter in the brain is glutamate, which binds to several receptors, including the N-methyl-D-aspartate receptor (NMDAR). NMDARs play roles in synaptic plasticity and excitotoxic cell death. Mitochondria, in turn, have fundamental functions such as ATP production and calcium regulation. Apoptosis, or the opening of the mitochondrial transition pore, resulting from disturbed mitochondrial calcium homeostasis, can lead to neuronal death. This undesirable outcome is related to the pathogenesis of diseases such as AD, PD, stroke, or traumatic brain injury. Therefore, the role of glutamate is indispensable for synaptic plasticity and cell death in relation to NMDA receptors, which are permeable to calcium and generate different signalling pathways depending on their location [88]. Calcium signalling is crucial for mitochondrial activity, and its dysregulation is linked to the development of neurodegenerative diseases such as AD. Dysregulated calcium homeostasis disrupts mitochondrial function, which is associated with neurodegeneration.8. Reactive Oxygen Species and Mitochondrial Dysfunction
ROS are chemically reactive molecules derived from oxygen metabolism that play crucial roles in cell signalling and homeostasis. Nevertheless, their excessive production can lead to oxidative stress, causing damage to cellular components, including proteins, lipids, and DNA [89]. Mitochondria, the energy-producing organelles within cells, are a significant source and target of ROS because of aerobic respiration and oxidative phosphorylation (OXPHOS) processes. Hence, recent studies have revealed that mitochondria are primarily responsible for generating significant amounts of ROS, like superoxide anion, oxygen free radicals, hydroxyl radicals, and hydrogen peroxide, through the mitochondrial electron transport chain (ETC) [90]. Consequently, these ROS may exacerbate oxidative stress, leading to cellular damage [91]. Moreover, other cellular sources of ROS include peroxisomes, endoplasmic reticulum, and phagocytic cells [92][93][94]. To counteract ROS-induced damage, cells have evolved an intricate antioxidant defense system. This system comprises antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, which scavenge and neutralise ROS. Additionally, non-enzymatic antioxidants, including vitamin C, vitamin E, and glutathione, play essential roles in maintaining cellular redox balance [95][96][97][98][99][100]. In this line, mitochondrial dysfunction, which implies an inability to overcome this oxidative stress, has been implicated in various diseases, including neurodegenerative disorders. In Alzheimer’s disease, specific histopathological features characterise brain tissue. These hallmarks include intracellular neurofibrillary tangles and extracellular senile plaques composed of the amyloid-beta peptide in an aggregated form, often associated with metal ions such as copper, iron, or zinc [101][102][103][104][105]. These metal ions can play a role in the generation of ROS when bound to the amyloid-beta peptide. When redox-active metal ions like copper or zinc bind to the amyloid-beta peptide, they facilitate the production of ROS [106]. The most reactive ROS produced in this process is the hydroxyl radical. Consequently, it may cause oxidative damage not only to the amyloid-beta peptide itself but also to surrounding molecules, including proteins, lipids, and other cellular components [107]. Regarding PD, it is well known how its pathophysiology involves a reduction of dopamine due to the impairment of dopamine-producing neurons in the substantia nigra [108]. Nevertheless, it has been proposed that another protein, alpha-synuclein, may also be associated with the pathophysiology of Parkinson’s disease. In this line, the previous literature suggested how mutations in a gene responsible for alpha synuclein production led to the formation of a mutant protein, which has the ability to induce the accumulation of dopamine in the cytoplasm of neurons [109][110]. Moreover, this mutant alpha-synuclein disrupts the integrity of the vesicles that contain dopamine, resulting in their permeation and subsequent leakage of dopamine into the cytoplasm. Therefore, the released dopamine suffers autoxidation, leading to the generation of hydrogen peroxide, superoxide molecules, and toxic dopamine-quinone species, promoting an oxidative environment [111], which can contribute to cellular damage and compromise Parkinson’s development.9. Inflammation and Mitochondrial Impairment in Brain Disease
The human brain represents the pinnacle of evolutionary progress due to its intricate functionality, enabling cognitive processes, memory retention, motor control, and emotional experiences. The primary objective of maintaining a healthy lifestyle is to ensure optimal cognitive function of the brain throughout the entirety of one’s lifespan. The incidence of neurological disorders and the challenges associated with maintaining optimal brain health escalate concomitantly with the advancing age of the populace. There have been numerous neurological conditions that have been associated with compromised cerebral function and adverse health consequences in individuals [112]. There are three main categories to categorise neurological diseases that lead to cognitive impairment (Figure 3). Firstly, brain diseases that cause obvious damage to brain structures include stroke, head trauma, malignant brain tumours, meningitis, and a variety of communication and sensory abnormalities. Brain disorders (such as PD and AD) and mental disorders (such as schizophrenia, depression, bipolar disorder, alcoholism, and drug misuse) are examples of functional brain disorders with a demonstrable breakdown of brain connections or networks. Finally, migraines, sleep difficulties, and other neurological conditions that do not cause obvious damage to the brain [112]. Mitochondrial dysfunction can be both the cause and consequence of inflammatory processes and lead to metabolic adaptations that can be either protective or progressively detrimental to our brains and lead to any of the above processes and diseases [113].
10. Mitochondria and Neurotransmitter Systems in Psychiatric Illnesses
11. Mitochondrial Biomarkers in Brain Disease
11.1. Mitochondrial Biomarkers in Neurodegenerative Disorders
The search for mitochondrial biomarkers in brain disease has predominantly focused on neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. These disorders are characterised by the accumulation of abnormal protein aggregates, mitochondrial dysfunction, and progressive neurodegeneration. In this line, neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, are characterised by progressive neuronal loss and cognitive decline. Mounting evidence suggests that mitochondrial dysfunction plays a critical role in the pathogenesis of these disorders, leading to a heightened interest in the search for mitochondrial biomarkers [133]. In Alzheimer’s disease, the measurement of mitochondrial biomarkers in cerebrospinal fluid (CSF), such as levels of amyloid-beta, tau, and phosphorylated tau proteins, has shown promise in aiding early diagnosis and tracking disease progression. Increased CSF levels of amyloid-beta and tau proteins, which are involved in the formation of plaques and neurofibrillary tangles in AD, correlate with mitochondrial dysfunction and neuronal damage. Neuroimaging techniques have also been employed to assess mitochondrial function in neurodegenerative disorders. Positron emission tomography (PET) imaging using radiotracers specific to mitochondrial function, such as [^18F]fluorodeoxyglucose (FDG), can measure glucose metabolism, which is closely linked to mitochondrial activity. Reduced glucose metabolism in specific brain regions affected by neurodegeneration can serve as an indirect marker of mitochondrial dysfunction.11.2. Mitochondrial Biomarkers in Psychiatric Illnesses
Psychiatric illnesses, including major depressive disorder, bipolar disorder, and schizophrenia, are complex and multifactorial conditions that significantly impact the well-being and quality of life of affected individuals. While these disorders have traditionally been associated with neurotransmitter imbalances and abnormalities in brain circuitry, emerging evidence suggests that mitochondrial dysfunction plays a crucial role in their pathogenesis. The identification and characterisation of mitochondrial biomarkers in psychiatric illnesses have garnered considerable attention in recent years, offering new insights into disease mechanisms, facilitating early diagnosis, and guiding personalised treatment approaches [134]. In major depressive disorder, alterations in mitochondrial function have been observed, leading to the exploration of mitochondrial biomarkers as potential diagnostic tools and treatment response predictors. Peripheral levels of mitochondrial DNA (mtDNA) have shown promise as biomarkers for differentiating subtypes of depression, monitoring treatment efficacy, and predicting outcomes. Additionally, markers of oxidative stress and mitochondrial enzyme activity have been investigated, providing further insights into mitochondrial dysfunction in depression [135]. Bipolar disorder, characterised by cyclic mood swings between manic and depressive episodes, has also been associated with mitochondrial dysfunction. Mitochondrial biomarkers, including mtDNA levels and markers of oxidative stress, have demonstrated potential for distinguishing between different mood states, predicting relapse, and monitoring treatment response. These biomarkers offer the possibility of developing targeted interventions that directly address mitochondrial dysfunction in bipolar disorder. Schizophrenia, a complex psychiatric disorder, is characterised by disturbances in perception, cognition, and emotional regulation. Growing evidence suggests that mitochondrial dysfunction contributes to the pathogenesis of schizophrenia, and mitochondrial biomarkers have been investigated to improve our understanding of the disorder. Peripheral levels of mtDNA, markers of oxidative stress, and mitochondrial enzyme activity have been explored as potential diagnostic markers and indicators of disease severity. These biomarkers may help identify specific subgroups of patients with distinct mitochondrial profiles, facilitating the development of tailored treatment strategies [122]. The identification and validation of reliable mitochondrial biomarkers in psychiatric illnesses face several challenges. Standardisation of sample collection and measurement techniques, as well as the establishment of reference ranges and diagnostic thresholds, are crucial for the widespread use and interpretation of these biomarkers. Longitudinal studies with large cohorts are needed to validate their diagnostic and prognostic value, monitor treatment response, and evaluate their potential for guiding personalised interventions. In conclusion, the investigation of mitochondrial biomarkers in psychiatric illnesses holds promise for advancing our understanding of disease mechanisms, improving early detection and diagnosis, and guiding personalised treatment approaches. Moreover, recent studies have highlighted the involvement of mitochondrial dynamics in psychiatric illnesses. Mitochondrial fusion and fission, processes that regulate mitochondrial morphology and distribution, have been found to be dysregulated in major depressive disorder and bipolar disorder. Altered expression and activity of proteins involved in mitochondrial dynamics, such as dynamin-related protein 1 (Drp1) and mitofusins, have been observed in postmortem brain tissue of individuals with these psychiatric conditions. These findings suggest that disruptions in mitochondrial dynamics contribute to the pathophysiology of these disorders and may represent potential therapeutic targets [136].12. Therapeutic Strategies: Mitochondrial Protective Agents
Mitochondrial damage refers to the alteration or dysfunction of mitochondria, the intracellular structures responsible for producing energy in the form of ATP. Mitochondria are vital for cellular functioning and play a key role in many biological processes. Some mechanisms are involved in mitochondrial damage. One of them is oxidative stress, associated with an imbalance between reactive oxygen species (ROS) production and elimination damaging mitochondrial structures and compromising their function [137]. Oxidative stress is associated with neurodegenerative diseases such as Alzheimer’s and Parkinson’s [138], as well as cardiovascular diseases, diabetes, and cancer [139][140][141]. In addition, Electron transport chain dysfunction leads to inadequate ATP production and increased ROS release, further damaging mitochondria [142]. Moreover, accumulated mitochondrial DNA damage can negatively affect mitochondrial function and contribute to ageing and various age-related diseases, such as cardiovascular diseases and neurodegenerative disorders [143]. A number of pharmaceutical medications and naturally occurring substances, which have been classified as mitochondrial protective agents, have demonstrated promise in the preservation of mitochondrial integrity and functionality. Mitochondrial damage-associated disorders may potentially benefit from the use of these interventions for treatment or prevention purposes. Regarding the evidence for these mitochondrial protective agents, preclinical studies have shown that coenzyme Q10 supplementation improves mitochondrial function, reduces oxidative stress, and provides neuroprotective effects in animal models of neurodegenerative diseases [144][145][146]. Coenzyme Q10 has antioxidant and anti-inflammatory effects and plays a role in energy production and mitochondrial stabilisation, which are mechanisms by which coenzyme Q10 exerts its neuroprotective effects [147]. Resveratrol is recognised for its antioxidant, anti-inflammatory, anti-apoptotic, and anticancer properties. It has been shown to influence mitochondrial function, redox biology, and dynamics in both in vitro and in vivo experimental models. Additionally, resveratrol can mitigate mitochondrial damage caused by specific stressors by enhancing mitochondria-located antioxidant enzymes, thereby reducing reactive species production in these organelles. Furthermore, resveratrol stimulates mitochondrial biogenesis, leading to an improved bioenergetic status of mammalian cells [148]. Regarding the effectiveness of curcumin on neurological diseases, some pharmacological effects have been attributed to it: anticancer, anti-inflammatory, antioxidant, antithrombotic, chemo sensitising and chemo preventive, antiatherosclerosis and cardioprotective, lipid-modifying, antibacterial, antifungal, antiviral, analgesic, pulmonoprotective, antidepressant, and antirheumatic activities [149][150][151][152][153][154]. A deficiency of L-carnitine can lead to impaired mitochondrial function and cellular metabolic alterations, potentially underlying several disease states [155]. Extensive preclinical and clinical research has confirmed the beneficial role of L-carnitine treatment in conditions such as myalgic encephalomyelitis, chronic fatigue syndrome [156], and neurodegenerative diseases [157]. Recent studies are beginning to unveil L-carnitine’s ability to modulate gene expression and other vital biological processes, in addition to its crucial role in mitochondrial energy metabolism [158][159]. Moreover, both L-carnitine and acetyl-L-carnitine have been linked to the prevention of toxic effects caused by beta-amyloid and the improvement of symptoms in Alzheimer’s disease [155]. These neuroprotective effects may be related to the reduction of amyloid-related mitochondrial dysfunction and a decrease in reactive oxygen species levels [160]. Acetyl-L-carnitine has shown promise in reducing beta-amyloid 1-42-induced protein and lipid oxidation, enhancing antioxidant potential by increasing glutathione and heat shock proteins [161], and preventing ATP depletion induced by beta-amyloid [162]. Experimental studies have demonstrated that treating cortical neurons with acetyl-L-carnitine can counter the neurotoxic effects of beta-amyloid 25–35 fragment [160] and attenuate beta-amyloid 1-42-induced toxicity and apoptosis, which could be attributed to a reduction in protein and lipid oxidation [161].13. Metabolic Modulators and Mitochondrial Function in Brain Diseases
Mitochondria, the most fascinating cell organelles, are crucial for cellular respiration and energy production. Additionally, they play a vital role in regulating cellular homeostasis by controlling energy production, calcium signalling, cell metabolism, and apoptosis. Mitochondrial dysfunction has been linked to a wide range of diseases, including neurodegenerative disorders, metabolic syndromes, and cardiovascular disease [163]. Mitochondrial dysfunction is a common feature of many diseases, including neurodegenerative disorders. Specifically, glucose metabolism dysregulation reduces energy generation in the brain and negatively impacts neuronal function; lipid metabolism alteration increased oxidative stress and accumulation of waste products; and dysregulation of calcium homeostasis ultimately contributes to disease pathogenesis [164][165]. The aetiology and pathogenesis of some neurodegenerative disorders and brain diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease exhibit a complex interaction between metabolism and mitochondrial function. Mitochondrial dysfunction and metabolic disorders can be key factors in the pathogenesis and progression of these diseases, highlighting the importance of further research and understanding of this relationship to develop effective therapeutic approaches to address these conditions. Specifically, in Alzheimer’s, there is an abnormal accumulation of beta-amyloid and tau proteins in the brain, leading to the formation of plaques and neurofibrillary tangles [166]. These protein aggregates can impair mitochondrial function and generate oxidative stress, resulting in neuronal damage and reduced energy production in mitochondria [167]. Several metabolic modulators have been studied in relation to mitochondrial function in the brain. These compounds have the ability to influence cellular metabolism and mitochondrial activity, which can have beneficial effects on brain function and the management of neurodegenerative diseases [168]. Regarding clinical evidence of metabolic modulators, preclinical and clinical studies have investigated their effectiveness in improving mitochondrial function and managing brain diseases. Some promising results have been reported. Specifically, preclinical studies have shown that alpha-lipoic acid improves mitochondrial function and reduces oxidative stress in the brain, which could be beneficial in neurodegenerative diseases [169][170][171][172]. Moreover, clinical trial studies using supplementation with alpha-lipoic acid have found that it improves cognition and mitochondrial function in patients with brain diseases [173][174][175].14. Gene Therapy for Restoring Mitochondrial Function
Gene therapy has emerged as a promising approach for targeting mitochondrial dysfunction in the context of brain disease. By introducing therapeutic genes into affected cells, gene therapy aims to restore mitochondrial function and mitigate the underlying pathological mechanisms. Several studies have demonstrated the potential of gene therapy in preclinical models of brain disease, highlighting its efficacy in enhancing mitochondrial biogenesis, improving oxidative phosphorylation, and attenuating neuroinflammation [176][177]. One promising gene therapy strategy involves the delivery of genes encoding mitochondrial-targeted antioxidants, such as manganese superoxide dismutase (MnSOD) or catalase, to counteract the elevated reactive oxygen species (ROS) levels observed in brain disease [178]. Another gene therapy approach involves the delivery of genes encoding mitochondrial fusion proteins, such as mitofusin 1 and 2 (MFN1 and MFN2), to enhance mitochondrial dynamics and restore impaired mitochondrial networks. Restoration of mitochondrial fusion has been shown to ameliorate neuronal damage and improve motor function in animal models of neurodegenerative disorders [179]. Additionally, advancements in genome editing technologies, particularly CRISPR-Cas9, have opened new possibilities for correcting mitochondrial DNA (mtDNA) mutations associated with brain diseases. Several studies have demonstrated the successful use of CRISPR-Cas9 in editing mtDNA to correct pathogenic mutations in patient-derived cells, providing a promising avenue for the treatment of mitochondrial diseases [180]. This technology offers a simple design, low cost, high efficiency, and the ability to simultaneously edit multiple loci without the need for plasmids [181]. The potential of CRISPR-Cas9 in correcting mtDNA mutations highlights its significance in the field of gene editing and its application in addressing genetic abnormalities associated with mitochondrial dysfunction.References
- San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782.
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723.
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Tornero-Aguilera, J.F.; Ruisoto, P.; Mielgo-Ayuso, J. Inflammation in COVID-19 and the Effects of Non-Pharmacological Interventions during the Pandemic: A Review. Int. J. Mol. Sci. 2022, 23, 15584.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080.
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044.
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261.
- Barros, M.H.; McStay, G.P. Modular biogenesis of mitochondrial respiratory complexes. Mitochondrion 2019, 50, 94–114.
- Zhou, Q.; Zhai, Y.; Lou, J.; Liu, M.; Pang, X.; Sun, F. Thiabendazole inhibits ubiquinone reduction activity of mitochondrial respiratory complex II via a water molecule mediated binding feature. Protein Cell 2011, 2, 531–542.
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514.
- Belhadj Slimen, I.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523.
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and Beta-Amyloid. Different Targets, Same Players: Calcium, Free Radicals and Mitochondria in the Mechanism of Neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115.
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6.
- Juárez, D.; González-Ravé, J.M.; Rubio-Arias, J.Á.; Clemente Suárez, V.J.; Valencia, M.; Abian Vicente, J. Isokinetic leg strength and power in elite handball players. J. Hum. Kinet. 2014, 41, 227–233.
- Bruni, F. Mitochondria: From Physiology to Pathology. Life 2021, 11, 991.
- Green, D.R. Apoptotic pathways: Paper wraps stone blunts scissors. Cell 2000, 102, 1–4.
- Puthalakath, H.; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002, 9, 505–512.
- Pellegrini, L.; Passer, B.J.; Tabaton, M.; Ganjei, J.K.; D’Adamio, L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999, 274, 21011–21016.
- van den Heuvel, M.P.; Sporns, O. Network hubs in the human brain. Trends Cogn. Sci. 2013, 17, 683–696.
- Pino, A.; Fumagalli, G.; Bifari, F.; Decimo, I. New neurons in adult brain: Distribution, molecular mechanisms and therapies. Biochem. Pharmacol. 2017, 141, 4–22.
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84.
- Kaminsky, N.; Bihari, O.; Kanner, S.; Barzilai, A. Connecting Malfunctioning Glial Cells and Brain Degenera-tive Disorders. Genom. Proteom. Bioinform. 2016, 14, 155–165.
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211.
- Yang, S.; Qin, C.; Hu, Z.-W.; Zhou, L.-Q.; Yu, H.-H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.-J.; Tian, D.-S. Microglia Reprogram Metabolic Profiles for Phenotype and Function Changes in Central Nervous System. Neurobiol. Dis. 2021, 152, 105290.
- Ganat, Y.M.; Silbereis, J.; Cave, C.; Ngu, H.; Anderson, G.M.; Ohkubo, Y.; Ment, L.R.; Vaccarino, F.M. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J. Neurosci. 2006, 26, 8609–8621.
- Fakhoury, M. Immune-mediated processes in neurodegeneration: Where do we stand? J. Neurol. 2016, 263, 1683–1701.
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735.
- Martin, J.B. Molecular Basis of the Neurodegenerative Disorders. N. Engl. J. Med. 1999, 340, 1970–1980.
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Ap-proaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851.
- Clemente-Suárez, V.J.; Ramos-Campo, D.J.; Mielgo-Ayuso, J.; Dalamitros, A.A.; Nikolaidis, P.A.; Hormeño-Holgado, A.; Tornero-Aguilera, J.F. Nutrition in the Actual COVID-19 Pandemic. A Narrative Review. Nutrients 2021, 13, 1924.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coac-tivator PGC-1. Cell 1999, 98, 115–124.
- Chew, K.; Zhao, L. Interactions of Mitochondrial Transcription Factor A with DNA Damage: Mechanistic Insights and Functional Implications. Genes 2021, 12, 1246.
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313.
- Chen, S.-D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ische-mia. Int. J. Mol. Sci. 2011, 12, 7199–7215.
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029.
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53.
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115.
- Kim, S.; Pajarillo, E.; Nyarko-Danquah, I.; Aschner, M.; Lee, E. Role of Astrocytes in Parkinson’s Disease Asso-ciated with Genetic Mutations and Neurotoxicants. Cells 2023, 12, 622.
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508.
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835.
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549.
- Pohland, M.; Pellowska, M.; Asseburg, H.; Hagl, S.; Reutzel, M.; Joppe, A.; Berressem, D.; Eckert, S.H.; Wurglics, M.; Schubert-Zsilavecz, M.; et al. MH84 improves mitochondrial dysfunction in a mouse model of early Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 18.
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767.
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; De Waal, R.M.W.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328.
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochon-drially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372.
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32.
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2017, 25, 24–34.
- MacDonald, M.E.; Barnes, G.; Srinidhi, J.; Duyao, M.P.; Ambrose, C.M.; Myers, R.H.; Gray, J.; Conneally, P.M.; Young, A.; Penney, J. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Genet. 1993, 30, 982–986.
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362.
- Johri, A.; Chandra, A.; Flint Beal, M. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46.
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.V.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra97.
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268.
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680.
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106.
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188.
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784.
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240.
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25.
- Hahn, A.; Zuryn, S. Mitochondrial Genome (mtDNA) Mutations that Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392.
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980.
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen. Med. 2020, 5, 22.
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237.
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639.
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660.
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267.
- Hamblet, N.S.; Castora, F.J. Elevated levels of the Kearns–Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutat. Res. 1997, 379, 253–262.
- Sequeira, A.; Martin, M.V.; Rollins, B.; Moon, E.A.; Bunney, W.E.; Macciardi, F.; Lupoli, S.; Smith, E.N.; Kelsoe, J.; Magnan, C.N.; et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet. 2012, 3, 103.
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37.
- Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Hung, P.-L.; Hsu, M.-H.; Wang, T.-J.; Wu, T.-Y.; Lee, T.-H.; Huang, Z.-D.; Chang, P.-C.; et al. Oxidative Insults and Mitochondrial DNA Mutation Promote Enhanced Autophagy and Mitophagy Compromising Cell Viability in Pluripotent Cell Model of Mitochondrial Disease. Cells 2019, 8, 65.
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608.
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322.
- Garrido-Pérez, N.; Vela-Sebastián, A.; López-Gallardo, E.; Emperador, S.; Iglesias, E.; Meade, P.; Jiménez-Mallebrera, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome. Int. J. Mol. Sci. 2020, 21, 3374.
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242.
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491.
- Yuan, X.; Liu, Y.; Bijonowski, B.M.; Tsai, A.-C.; Fu, Q.; Logan, T.M.; Ma, T.; Li, Y. NAD(+)/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun. Biol. 2020, 3, 774.
- Clemente-Suárez, V.J.; Robles-Pérez, J.J. Psycho-physiological response of soldiers in urban combat. An. Psicol. 2013, 29, 598–603.
- Van der Velpen, V.; Teav, T.; Gallart-Ayala, H.; Mehl, F.; Konz, I.; Clark, C.; Oikonomidi, A.; Peyratout, G.; Henry, H.; Delorenzi, M.; et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 93.
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090.
- Xie, H.-R.; Hu, L.-S.; Li, G.-Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092.
- Agholme, L.; Lindström, T.; Kågedal, K.; Marcusson, J.; Hallbeck, M. An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 2010, 20, 1069–1082.
- Devine, M.J.; Birsa, N.; Kittler, J.T. Miro sculpts mitochondrial dynamics in neuronal health and disease. Neurobiol. Dis. 2016, 90, 27–34.
- Avdoshina, V.; Fields, J.A.; Castellano, P.; Dedoni, S.; Palchik, G.; Trejo, M.; Adame, A.; Rockenstein, E.; Eugenin, E.; Masliah, E.; et al. The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotox. Res. 2016, 29, 583–593.
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55.
- Crews, L.; Tsigelny, I.; Hashimoto, M.; Masliah, E. Role of synucleins in Alzheimer’s disease. Neurotoxicity Res. 2009, 16, 306–317.
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430.
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal 2011, 14, 459–468.
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Sarniak, A.; Lipińska, J.; Tytman, K.; Lipińska, S. Endogenous mechanisms of reactive oxygen species (ROS) generation. Postep. Hig. Med. Dosw. Online 2016, 70, 1150–1165.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303.
- Clemente-Suárez, V.J.; Mielgo-Ayuso, J.; Quiles, J.L.; Varela-Lopez, A.; Aranda, P. Effect of α-tocopherol megadoses on hematologic parameters and antioxidant capacity of rats in an ultraendurance probe. Physiol. Int. 2017, 104, 291–300.
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298.
- García-Giménez, J.L.; Pallardó, F.V. Maintenance of glutathione levels and its importance in epigenetic regulation. Front. Pharmacol. 2014, 5, 88.
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2015.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal 2020, 32, 677–700.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52.
- Tõugu, V.; Palumaa, P. Coordination of zinc ions to the key proteins of neurodegenerative diseases: Aβ, APP, α-synuclein and PrP. Coord. Chem. Rev. 2012, 256, 2219–2224.
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206.
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128.
- Wang, P.; Wang, Z.-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290.
- Tiiman, A.; Palumaa, P.; Tõugu, V. The missing link in the amyloid cascade of Alzheimer’s disease—Metal ions. Neurochem. Int. 2013, 62, 367–378.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 95–121.
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942.
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643.
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 11869–11874.
- Wang, Y.; Pan, Y.; Li, H. What is brain health and why is it important? BMJ 2020, 371, m3683.
- Currais, A. Ageing and inflammation—A central role for mitochondria in brain health and disease. Ageing Res. Rev. 2015, 21, 30–42.
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937.
- Yan, F.; Tang, H.; Wang, L.; Huang, L.; Zhang, J. Editorial: Mitochondrial Dysfunction in Stroke. Front. Aging Neurosci. 2022, 14, 888952.
- Widmann, C.N.; Heneka, M.T. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014, 13, 630–636.
- Zsurka, G.; Kunz, W.S. Mitochondrial involvement in neurodegenerative diseases. IUBMB Life 2013, 65, 263–272.
- Chan, S.T.; McCarthy, M.J.; Vawter, M.P. Psychiatric drugs impact mitochondrial function in brain and other tissues. Schizophr. Res. 2020, 217, 136–147.
- Spiers, J.G.; Chen, H.-J.C.; Bourgognon, J.-M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free. Radic. Biol. Med. 2019, 134, 468–483.
- Foyet, S.H.; Balmus, I.-M.; Hervé, N.A.H.; Emmanuel, A.A.; Guenne, S.; Kiendrebéogo, M.; Ciobica, A. Ethnopharmacological approaches in mood and anxiety disorders. The relevance of the oxidative stress status. J. Complement. Integr. Med. 2017, 14.
- Vaváková, M.; Ďuračková, Z.; Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid. Med. Cell. Longev. 2015, 2015, 898393.
- Madireddy, S.; Madireddy, S. Therapeutic Interventions to Mitigate Mitochondrial Dysfunction and Oxidative Stress-Induced Damage in Patients with Bipolar Disorder. Int. J. Mol. Sci. 2022, 23, 1844.
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 8881770.
- Kato, T.A.; Yamauchi, Y.; Horikawa, H.; Monji, A.; Mizoguchi, Y.; Seki, Y.; Hayakawa, K.; Utsumi, H.; Kanba, K. Neurotransmitters, psychotropic drugs and microglia: Clinical implications for psychiatry. Curr. Med. Chem. 2013, 20, 331–344.
- Ye, N.; Song, Z.; Zhang, A. Dual ligands targeting dopamine D2 and serotonin 5-HT1A receptors as new antipsychotical or anti-Parkinsonian agents. Curr. Med. Chem. 2013, 21, 437–457.
- Clemente-Suárez, V.J. Multidisciplinary intervention in the treatment of mixed anxiety and depression disorder. Physiol. Behav. 2020, 219, 112858.
- Lopresti, A.L.; Hood, S.D.; Drummond, P.D. A review of lifestyle factors that contribute to important pathways associated with major depression: Diet, sleep and exercise. J. Affect. Disord. 2013, 148, 12–27.
- Sacher, J.; Rekkas, P.V.; Wilson, A.A.; Houle, S.; Romano, L.; Hamidi, J.; Rusjan, P.; Fan, I.; Stewart, D.E.; Meyer, J.H. Relationship of monoamine oxidase—A distribution volume to postpartum depression and postpartum crying. Neuropsychopharmacology 2015, 40, 429–435.
- Clemente-Suárez, V.J.; Bustamante-Sanchez, Á.; Mielgo-Ayuso, J.; Martínez-Guardado, I.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Antioxidants and Sports Performance. Nutrients 2023, 15, 2371.
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1049–1069.
- Francis, P.T. Glutamatergic systems in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S15–S21.
- Bemeur, C. Oxidative Stress in the Central Nervous System Complications of Chronic Liver Failure. In Studies on Hepatic Disorders; Springer: Berlin/Heidelberg, Germany, 2015; pp. 357–370.
- Tassone, A.; Meringolo, M.; Ponterio, G.; Bonsi, P.; Schirinzi, T.; Martella, G. Mitochondrial Bioenergy in Neurodegenerative Disease: Huntington and Parkinson. Int. J. Mol. Sci. 2023, 24, 7221.
- Mandal, P.K.; Gaur, S.; Roy, R.G.; Samkaria, A.; Ingole, R.; Goel, A. Schizophrenia, Bipolar and Major Depressive Disorders: Overview of Clinical Features, Neurotransmitter Alterations, Pharmacological Interventions, and Impact of Oxidative Stress in the Disease Process. ACS Chem. Neurosci. 2022, 13, 2784–2802.
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68.
- Khadimallah, I.; Jenni, R.; Cabungcal, J.-H.; Cleusix, M.; Fournier, M.; Beard, E.; Klauser, P.; Knebel, J.-F.; Murray, M.M.; Retsa, C.; et al. Mitochondrial, exosomal miR137-COX6A2 and gamma synchrony as biomarkers of parvalbumin interneurons, psychopathology, and neurocognition in schizophrenia. Mol. Psychiatry 2022, 27, 1192–1204.
- Carreira Míguez, M.; Clemente Suárez, V.J. Physical activity levels affect mental health and behavior in men. J. Men’s Health 2023, 1, 12.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127.
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178.
- Li, Z.-Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun. 2011, 414, 5–8.
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Yang, X.; Dai, G.; Li, G.; Yang, E.S. Coenzyme Q10 reduces β-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2010, 41, 110–113.
- Fouad, G.I. Combination of Omega 3 and Coenzyme Q10 Exerts Neuroprotective Potential Against Hypercholesterolemia-Induced Alzheimer’s-Like Disease in Rats. Neurochem. Res. 2020, 45, 1142–1155.
- Komaki, H.; Faraji, N.; Komaki, A.; Shahidi, S.; Etaee, F.; Raoufi, S.; Mirzaei, F. Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 147, 14–21.
- Bagheri, S.; Haddadi, R.; Saki, S.; Kourosh-Arami, M.; Rashno, M.; Mojaver, A.; Komaki, A. Neuroprotective effects of coenzyme Q10 on neurological diseases: A review article. Front. Neurosci. 2023, 17, 1188839.
- Jardim, F.R.; De Rossi, F.T.; Nascimento, M.X.; Barros, R.G.d.S.; Borges, P.A.; Prescilio, I.C.; De Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101.
- Alikiaii, B.; Bagherniya, M.; Askari, G.; Sathyapalan, T.; Sahebkar, A. Evaluation of the effect of curcumin on pneumonia: A systematic review of preclinical studies. Phytotherapy Res. 2021, 35, 1939–1952.
- Bagherniya, M.; Nobili, V.; Blesso, C.N.; Sahebkar, A. Medicinal plants and bioactive natural compounds in the treatment of non-alcoholic fatty liver disease: A clinical review. Pharmacol. Res. 2018, 130, 213–240.
- Ghandadi, M.; Sahebkar, A. Curcumin: An Effective Inhibitor of Interleukin-6. Curr. Pharm. Des. 2017, 23, 921–931.
- Mollazadeh, H.; Cicero, A.F.G.; Blesso, C.N.; Pirro, M.; Majeed, M.; Sahebkar, A. Immune modulation by curcumin: The role of interleukin-10. Crit. Rev. Food Sci. Nutr. 2019, 59, 89–101.
- Panahi, Y.; Ahmadi, Y.; Teymouri, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a potential candidate for treating hyperlipidemia: A review of cellular and metabolic mechanisms. J. Cell. Physiol. 2018, 233, 141–152.
- Teymouri, M.; Pirro, M.; Johnston, T.P.; Sahebkar, A. Curcumin as a multifaceted compound against human papilloma virus infection and cervical cancers: A review of chemistry, cellular, molecular, and preclinical features. Biofactors 2017, 43, 331–346.
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717.
- Plioplys, A.V.; Plioplys, S. Serum levels of carnitine in chronic fatigue syndrome: Clinical correlates. Neuropsychobiology 1995, 32, 132–138.
- Latham, L.E.; Wang, C.; Patterson, T.A.; Slikker, W.; Liu, F. Neuroprotective Effects of Carnitine and Its Potential Application to Ameliorate Neurotoxicity. Chem. Res. Toxicol. 2021, 34, 1208–1222.
- Vidal-Casariego, A.; Burgos-Peláez, R.; Martínez-Faedo, C.; Calvo-Gracia, F.; Valero-Zanuy, M.; Luengo-Pérez, L.M.; Cuerda-Compés, C. Metabolic Effects of L-carnitine on Type 2 Diabetes Mellitus: Systematic Review and Meta-analysis. Exp. Clin. Endocrinol. Diabetes 2013, 121, 234–238.
- Förster, L.; Indra, D.; Rosenberger, K.; Zver, L.; Hofbauer, R. L-carnitine exerts a nutrigenomic effect via direct modulation of nuclear receptor signaling in adipocytes, hepatocytes and SKMC, demonstrating its nutritional impact. Nutr. Res. 2021, 85, 84–98.
- Virmani, M.; Caso, V.; Spadoni, A.; Rossi, S.; Russo, F.; Gaetani, F. The action of acetyl-L-carnitine on the neurotoxicity evoked by amyloid fragments and peroxide on primary rat cortical neurones. Ann. N. Y. Acad. Sci. 2001, 939, 162–178.
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 398–408.
- Dhitavat, S.; Ortiz, D.; Shea, T.B.; Rivera, E.R. Acetyl-L-carnitine protects against amyloid-beta neurotoxicity: Roles of oxidative buffering and ATP levels. Neurochem. Res. 2002, 27, 501–505.
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 8848.
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343.
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560.
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810.
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091.
- Arduino, D.M.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial metabolism modulation: A new therapeutic approach for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 105–119.
- Hagen, T.M.; Ingersoll, R.T.; Lykkesfeldt, J.; Liu, J.; Wehr, C.M.; Vinarsky, V.; Bartholomew, J.C.; Ames, B.N. (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 1999, 13, 411–418.
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959.
- Tai, S.; Zheng, Q.; Zhai, S.; Cai, T.; Xu, L.; Yang, L.; Jiao, L.; Zhang, C. Alpha-lipoic acid mediates clearance of iron accumulation by regulating iron metabolism in a Parkinson’s disease model induced by 6-OHDA. Front. Neurosci. 2020, 14, 612.
- Liu, L.; Yang, S.; Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP(+)-induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 2021, 45, 422–431.
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 38, 111–120.
- Hager, K.; Kenklies, M.; McAfoose, J.; Engel, J.; Münch, G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease—A 48 months follow-up analysis. J. Neural Transm. Suppl. 2007, 72, 189–193.
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. J. Neurodegener. Dis. 2013, 2013, 1–7.
- Smith, R.A.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352.
- Mounsey, R.B.; Teismann, P. Mitochondrial dysfunction in Parkinson’s disease: Pathogenesis and neuroprotection. Park. Dis. 2011, 2011, 617472.
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649.
- Silva-Pinheiro, P.; Minczuk, M. The potential of mitochondrial genome engineering. Nat. Rev. Genet. 2022, 23, 199–214.
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.-S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol. Ther. 2021, 29, 571–586.
- Zhu, Y. Advances in CRISPR/Cas9. BioMed Res. Int. 2022, 2022, 9978571.