Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natsuko Aida | -- | 1980 | 2023-09-11 11:35:08 | | | |
| 2 | Rita Xu | Meta information modification | 1980 | 2023-09-11 11:37:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Aida, N.; Saito, A.; Azuma, T. Next-Generation Sequencing in Bone Genetic Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/49015 (accessed on 28 July 2026).
Aida N, Saito A, Azuma T. Next-Generation Sequencing in Bone Genetic Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/49015. Accessed July 28, 2026.
Aida, Natsuko, Akiko Saito, Toshifumi Azuma. "Next-Generation Sequencing in Bone Genetic Diseases" Encyclopedia, https://encyclopedia.pub/entry/49015 (accessed July 28, 2026).
Aida, N., Saito, A., & Azuma, T. (2023, September 11). Next-Generation Sequencing in Bone Genetic Diseases. In Encyclopedia. https://encyclopedia.pub/entry/49015
Aida, Natsuko, et al. "Next-Generation Sequencing in Bone Genetic Diseases." Encyclopedia. Web. 11 September, 2023.
Copy Citation
The development of next-generation sequencing (NGS) has dramatically increased the speed and volume of genetic analysis. Furthermore, the range of applications of NGS is rapidly expanding to include genome, epigenome (such as DNA methylation), metagenome, and transcriptome analyses (such as RNA sequencing and single-cell RNA sequencing). NGS enables genetic research by offering various sequencing methods as well as combinations of methods. Bone tissue is the most important unit supporting the body and is a reservoir of calcium and phosphate ions, which are important for physical activity.
next-generation sequencing
genome
bone genetic diseases
1. Introduction
The world is now empowered by big data. In life science research, next-generation sequencing (NGS) has enabled the simultaneous reading of numerous fragmented DNA molecules and their sequencing in a massively parallel manner [1][2]. In 2003, the completion of the Human Genome Project was declared [3], and the Telomere-to-Telomere (T2T) consortium presented the complete sequence of the human genome, T2T-CHM13, in 2022 [4]. The release of all analyzed data has greatly expanded the research agenda. Using Sanger sequencing, the mainstream method at the time, the cost of analyzing an individual’s genome was approximately 10 USD million 20 years ago [5][6]. The promotion of the “Advanced Sequencing Technology Awards”, commonly known as the “1000 USD Genome Program”, has made it possible to bring the cost down to 1000 USD per person [7]. The ability to decode the human genome in a few days and its reduced cost has led many researchers to focus on using NGS to elucidate the genetic underpinnings of diseases [8]. Compared with conventional Sanger sequencing, NGS can yield an overwhelming amount of data in a short time [9].
Bone genetic disorder is a general term for diseases that cause abnormalities in the formation and maintenance of the skeleton because of defects in the growth, development, and differentiation of bone, cartilage, and other skeleton-forming tissues. Nosology and classification of genetic skeletal disorders 2023 revision for skeletal dysplasia lists 771 diseases, of which more than 90% are genetic diseases [10]. Many bone genetic diseases occur infrequently, but their causative genes are diverse. Although some causative genes have been elucidated, others remain unclear. However, investigating bone genetic diseases is valuable in that exploring the role of causative genes in human bone diseases will clarify their subsequent effect on developmental processes. Furthermore, the analysis of genetic mutations in many bone disorders will help develop new drugs and therapies, such as genome editing technology, for correcting mutated genes, especially in patients with genetic diseases that are difficult to treat [11][12].
2. Next-Generation Sequencing
The human genome has approximately 3.05 billion base pairs, and several methods can be employed for decoding them. The analysis method can be selected according to the purpose. Genomic analysis is one method used to decode the human genome. Epigenomic analysis deals with genetic information defined by chemical modifications of DNA or histone proteins without any changes in the DNA sequence. Transcriptome analysis comprehensively analyzes transcripts such as RNA transcribed from DNA [13] (Figure 1).

Figure 1. Various NGS-based sequencing methods.
Researchers categorized four major methods. The first is genomic sequencing, which comprehensively detects genomic mutations. The second is epigenomic sequencing, which examines the mode of genomic modifications involved in the regulation of gene expression, including DNA and histone modifications, chromatin conformation, and non-coding RNAs. The third is transcriptome sequencing, which comprehensively examines transcripts, and the fourth is metagenomic sequencing, which comprehensively sequences the DNA of all bacterial flora.
2.1. Genomics Analysis
Genomic analysis involves a comprehensive analysis of the human genome to determine the DNA sequences that make up the genome. Next-generation sequencers are commonly used to decipher the human genome, particularly disease-specific genomes. The primary purpose of genomic data analysis is to identify the genetic variants that cause the disease phenotype, including bone genetic disorders, from sequence data (Figure 2).
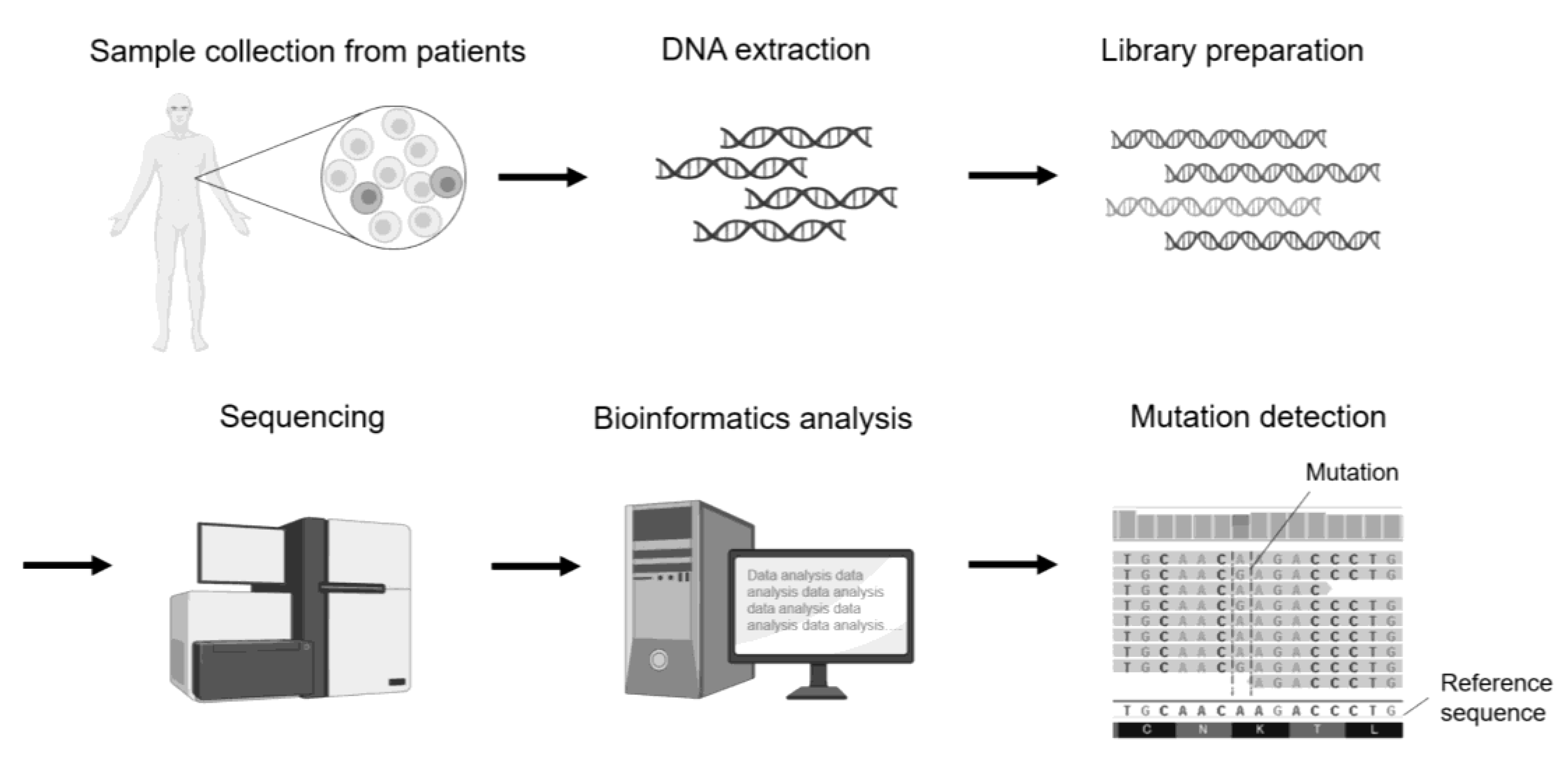
Figure 2. Schematic diagram of mutation analysis by genomics sequencing using a sample derived from a patient with a genetic disease.
A sample is collected from the blood or other tissues of patients with genetic diseases, and DNA is isolated. The DNA is used to prepare a library, which is then sequenced using a next-generation sequencer. Bioinformatics analysis is performed on the obtained sequence data, and mutations are identified by comparing them with reference sequences.
2.1.1. Advances in Genetic Analysis: From Gene Chips and Panels to Whole-Exome Sequencing (WES), and Finally, WGS
Even if a disease is assumed to be genetic, it is difficult to analyze genetic diseases with multiple causative genes using conventional Sanger sequencing, making the analysis more complex.
In general, the identification of a causative gene does not immediately lead to the elucidation of disease pathogenesis [14]. In some cases, various genetic abnormalities share a common pathology, such as muscular dystrophy. In the “Development of Human Muscle DNA Chip” all known cDNA information expressed in human skeletal and cardiac muscle was collected, and cDNA fragments of human skeletal and cardiac muscle were cloned in silico to eliminate cross-hybridization. The analysis of differences in gene expression between muscle diseases, the correlation between patients, and the signal transduction system analysis are considered promising in providing basic information for drug discovery.
Handling large amounts of raw data is difficult. Data collection over time and the identification of markers to evaluate the process of regeneration at the gene expression level will provide basic data for the future. The classification of related genes by cluster analysis may enable further pathophysiological analyses.
Patients with osteogenesis imperfecta (OI), a genetic disorder that causes repeated fractures, are clinically and genetically diverse. Mutations in COL1A1 or COL1A2 are causative in approximately 85–90% of cases. Mutations in IFITM5 [15], SERPINF1 [16], CRTAP [17], LEPRE1 [18], P3H1 [19], PPIB [20], SERPINH1 [21], FKBP10 [22], SP7 [23], BMP1 [24], TMEM38B [25], WNT1 [26], CREB3l1 [27], SPARC [28], FAM46a [29], MBTPS2 [30], MESD [31], SEC24d [32], CCDC134 [33], P4HB [34], PLOD2 [35], PLS3 [36], and KDELR2 [37] have also been reported. All these mutations affect type I collagen quality and quantity. A gene panel is an excellent tool for comprehensively analyzing and elucidating these mutations and is already commercially available [38]. In addition, the elucidation of pathological conditions by genotype–phenotype analysis is very important, and the use of gene panels for OI is becoming common. However, panel production and the additional discovery of new causative genes require a new set of panels, which is also costly. However, some unexpected genes may have been overlooked. This disadvantage can be circumvented by whole-exome sequencing (WES) [39]. The exome is a large panel of genes (approximately 45 Mb) that is enriched with all exons of the 20,000 genes known to date. The advantage of using WES is its potential to identify novel gene–disease associations. Most current genetic disease-causing gene searches use WES. However, the exome has gaps; for example, it only partially covers most introns. Therefore, a clear disadvantage of WES is its inability to identify deep intronic variants that can lead to disease-causing splicing abnormalities. The all-in-one solution for assessing coding and non-coding variants as well as structural variants (mainly copy number variants) is WGS. Future genetic analyses will likely converge rapidly with WGS analysis.
2.1.2. Panel Sequencing
Recently, cancer genome medicine has become available in cancer medicine. Cancer gene panel tests, which can examine multiple genes simultaneously, are expected to provide tailor-made genome therapies, such as selecting effective drugs through the genetic analysis of patients. Such targeted resequencing is not limited to cancer but is also effective in diagnosing genetic diseases [40]. Using PCR, libraries are constructed and sequenced for specific regions. Consequently, one can concentrate on individual genes and target specific regions in the genome. Many whole-genome analyses have accumulated information, and databases have been constructed. Several bone disease panels, such as the osteogenesis imperfecta, osteopetrosis sugar genetic, and skeletal disease panels, are commercially available. In 2021, Nakamura et al. developed a diagnostic gene panel for Gorlin syndrome caused by gain-of-function mutations in the hedgehog signaling pathway. The gene panel analyzed the PTCH1, PTCH2, SUFU, and SMO genes, all of which are associated with Gorlin syndrome. They reported the presence of simultaneous mutations in PTCH1 and PTCH2 in patients with Gorlin syndrome [41].
2.1.3. Whole-Exome Sequencing (WES)
Exome analysis, which decodes only the exonic regions, about 1.5% of the entire human genome, makes it possible to identify causative genes and nucleotide variants of inherited diseases [42]. Narrowing down the regions to be sequenced can reduce the time required to analyze an individual’s genome sequence. The decoded genome sequence is mapped using software, and only exonic regions with deletions and other genomic variants that cause genetic disease are extracted from the mapped file. In 2018, WES and panel sequencing analyses identified potential causal variants in 411 patients with skeletal dysplasia (288 families). Sharing phenotypic and genotypic data from a large molecularly characterized skeletal dysplasia cohort is expected to improve the diagnosis of these patients in the target population [43].
2.1.4. Whole-Genome Sequencing (WGS)
WGS is a comprehensive method that analyzes the entire genome. Genomic information is expected to be helpful in the diagnosis and identification of hereditary diseases, the characterization of somatic mutations caused by cancer, and the identification of effective drugs for diseases [44]. It is also optimal for identifying and establishing novel genome sequences. It is possible to obtain an overall picture of the genome using genomic analysis. Moreover, it searches for any mutations that might have been missed using a targeted approach. In this respect, WGS has significant advantages. In 2020, Andersson et al. conducted a genetic search using WGS to determine whether patients with osteogenesis imperfecta (OI) and mutations in COL1A1/A2 had other genetic variants that might affect tooth development. Several missense variants were found, suggesting that the additive effects of pathological variants in COL1A1, COL1A2, and CERB3L1 may be necessary for osteogenesis imperfecta [45].
2.2. Epigenomics Sequencing
Epigenetic analysis is essential for elucidating bone diseases. Epigenetics refers to the further modification of a gene. Humans are multicellular organisms comprising many differentiated cells with a single genome. Several mechanisms control the expression of cell-specific genes. Epigenetics are the mechanisms that regulate gene expression without changing the DNA sequence, including DNA methylation, histone modification (methylation, acetylation, phosphorylation, etc.), and chromatin remodeling [46]. Information on the epigenetics of bone cells is obtained from the ENCODE project (http://www.genome.gov/encode/) (accessed on 1 August 2023).
2.2.1. ChIP-Sequencing
Chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq), a combination of chromatin immunoprecipitation (ChIP) assays and sequencing, is a powerful method for identifying the DNA-binding sites of transcription factors and other proteins on a genome-wide scale. In histone modifications, acetylated histones, especially acetylation of K9 and K27 on histone H3 (H3K9ac, H3K27ac), lead to chromatin decondensation and readily allow transcription into RNA, while trimethylation on K9 and K27 of histone H3 (H3K9me3, H3K27me 3) inhibits transcription [47]. Histone modifications during osteoblast differentiation have been examined using Chip-seq. The epigenetic regulation of Runx2, a master regulator of bone differentiation, involves the enrichment of H3K4me3 and H3K27ac marks in the Runx2 P1 promoter region [48]. Ankylosing spondylitis (AS) is a rheumatic disease with pathological bone formation that causes bone ankylosis and deformity; Yu et al. reported a 31 upregulated gene (SNP-adjacent superfamily) highly implicated in promoting abnormal osteogenic differentiation by integration of SNP data from ChIP-seq, RNA-seq, and the NHGRI-EBI GWAS catalog [49].
2.2.2. ATAC-Sequencing
ATAC-seq analyzes only the sequences of open chromatin regions, which are genomic regions that are likely to be transcribed into RNA [50]. In ATAC-seq, cellular activation leads to an increase in accessible chromatin regions, resulting in enhanced transcriptional activity. Active genes adopt an open chromatin state, facilitating accessibility to transcription factors and RNA polymerase and leading to upregulated gene expression. Consequently, transcriptional products, including RNA (mRNA), increase [51]. Unlike ChIP-seq, this method is versatile as it does not require specific antibodies [52]. In 2018, Liu et al. performed ATAC-seq using articular knee cartilage from patients with osteoarthritis (OA), a common joint disease, and integrated the analysis with previously reported RNA-seq data from patients with OA. They confirmed that the promoters and enhancers of genes involved in OA pathogenesis are altered. These results suggest that aberrant enhancer usage is associated with mesenchymal stem cell (MSC) differentiation and chondrogenesis in OA [53].
References
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-generation sequencing technology: Current trends and advancements. Biology 2023, 12, 997.
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141.
- Vockley, J.; Aartsma-Rus, A.; Cohen, J.L.; Cowsert, L.M.; Howell, R.R.; Yu, T.W.; Wasserstein, M.P.; Defay, T. Whole-genome sequencing holds the key to the success of gene-targeted therapies. Am. J. Med. Genet. C Semin. Med. Genet. 2023, 193, 19–29.
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53.
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658.
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238.
- Schloss, J.A.; Gibbs, R.A.; Makhijani, V.B.; Marziali, A. Cultivating DNA sequencing technology after the human genome project. Annu. Rev. Genomics Hum. Genet. 2020, 21, 117–138.
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46.
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age; ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351.
- Unger, S.; Ferreira, C.R.; Mortier, G.R.; Ali, H.; Bertola, D.R.; Calder, A.; Cohn, D.H.; Cormier-Daire, V.; Girisha, K.M.; Hall, C.; et al. Nosology of genetic skeletal disorders: 2023 revision. Am. J. Med. Genet. A 2023, 191, 1164–1209.
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361.
- Marwaha, S.; Knowles, J.W.; Ashley, E.A. A guide for the diagnosis of rare and undiagnosed disease: Beyond the exome. Genome Med. 2022, 14, 23.
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59.
- Davis, E.E.; Katsanis, N. The ciliopathies: A transitional model into systems biology of human genetic disease. Curr. Opin. Genet. 2012, 22, 290–303.
- Semler, O.; Garbes, L.; Keupp, K.; Swan, D.; Zimmermann, K.; Becker, J.; Iden, S.; Wirth, B.; Eysel, P.; Koerber, F.; et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am. J. Hum. Genet. 2012, 91, 349–357.
- Becker, J.; Semler, O.; Gilissen, C.; Li, Y.; Bolz, H.J.; Giunta, C.; Bergmann, C.; Rohrbach, M.; Koerber, F.; Zimmermann, K.; et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011, 88, 362–371.
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006, 127, 291–304.
- Caparrós-Martin, J.A.; Valencia, M.; Pulido, V.; Martínez-Glez, V.; Rueda-Arenas, I.; Amr, K.; Farra, C.; Lapunzina, P.; Ruiz-Perez, V.L.; Temtamy, S.; et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. A 2013, 161A, 1354–1369.
- Cabral, W.A.; Chang, W.; Barnes, A.M.; Weis, M.; Scott, M.A.; Leikin, S.; Makareeva, E.; Kuznetsova, N.V.; Rosenbaum, K.N.; Tifft, C.J.; et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007, 39, 359–365.
- van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527.
- Christiansen, H.E.; Schwarze, U.; Pyott, S.M.; AlSwaid, A.; Al Balwi, M.; Alrasheed, S.; Pepin, M.G.; Weis, M.A.; Eyre, D.R.; Byers, P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 389–398.
- Alanay, Y.; Avaygan, H.; Camacho, N.; Utine, G.E.; Boduroglu, K.; Aktas, D.; Alikasifoglu, M.; Tuncbilek, E.; Orhan, D.; Bakar, F.T.; et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 551–559.
- Lapunzina, P.; Aglan, M.; Temtamy, S.; Caparrós-Martín, J.A.; Valencia, M.; Letón, R.; Martínez-Glez, V.; Elhossini, R.; Amr, K.; Vilaboa, N.; et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 87, 110–114.
- Pihlajaniemi, T.; Dickson, L.A.; Pope, F.M.; Korhonen, V.R.; Nicholls, A.; Prockop, D.J.; Myers, J.C. Osteogenesis imperfecta: Cloning of a pro-alpha 2(I) collagen gene with a frameshift mutation. J. Biol. Chem. 1984, 259, 12941–12944.
- Shaheen, R.; Alazami, A.M.; Alshammari, M.J.; Faqeih, E.; Alhashmi, N.; Mousa, N.; Alsinani, A.; Ansari, S.; Alzahrani, F.; Al-Owain, M.; et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J. Med. Genet. 2012, 49, 630–635.
- Keupp, K.; Beleggia, F.; Kayserili, H.; Barnes, A.M.; Steiner, M.; Semler, O.; Fischer, B.; Yigit, G.; Janda, C.Y.; Becker, J.; et al. Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 2013, 92, 565–574.
- Symoens, S.; Malfait, F.; D’hondt, S.; Callewaert, B.; Dheedene, A.; Steyaert, W.; Bächinger, H.P.; De Paepe, A.; Kayserili, H.; Coucke, P.J. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 2013, 8, 154.
- Mendoza-Londono, R.; Fahiminiya, S.; Majewski, J.; Care4Rare Canada Consortium; Tétreault, M.; Nadaf, J.; Kannu, P.; Sochett, E.; Howard, A.; Stimec, J.; et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 2015, 96, 979–985.
- Doyard, M.; Bacrot, S.; Huber, C.; Di Rocco, M.; Goldenberg, A.; Aglan, M.S.; Brunelle, P.; Temtamy, S.; Michot, C.; Otaify, G.A.; et al. FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J. Med. Genet. 2018, 55, 278–284.
- Lindert, U.; Cabral, W.A.; Ausavarat, S.; Tongkobpetch, S.; Ludin, K.; Barnes, A.M.; Yeetong, P.; Weis, M.; Krabichler, B.; Srichomthong, C.; et al. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat. Commun. 2016, 7, 11920.
- Moosa, S.; Yamamoto, G.L.; Garbes, L.; Keupp, K.; Beleza-Meireles, A.; Moreno, C.A.; Valadares, E.R.; de Sousa, S.B.; Maia, S.; Saraiva, J.; et al. Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta. Am. J. Hum. Genet. 2019, 105, 836–843.
- Garbes, L.; Kim, K.; Rieß, A.; Hoyer-Kuhn, H.; Beleggia, F.; Bevot, A.; Kim, M.J.; Huh, Y.H.; Kweon, H.S.; Savarirayan, R.; et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 2015, 96, 432–439.
- Dubail, J.; Brunelle, P.; Baujat, G.; Huber, C.; Doyard, M.; Michot, C.; Chavassieux, P.; Khairouni, A.; Topouchian, V.; Monnot, S.; et al. Homozygous Loss-of-Function Mutations in CCDC134 Are Responsible for a Severe Form of Osteogenesis Imperfecta. J. Bone Miner. Res. 2020, 35, 1470–1480.
- Li, L.; Zhao, D.; Zheng, W.; Wang, O.; Jiang, Y.; Xia, W.; Xing, X.; Li, M. A novel missense mutation in P4HB causes mild osteogenesis imperfecta. Biosci. Rep. 2019, 39, BSR20182118.
- Ha-Vinh, R.; Alanay, Y.; Bank, R.A.; Campos-Xavier, A.B.; Zankl, A.; Superti-Furga, A.; Bonafe, L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet. A 2004, 131, 115–120.
- Chen, T.; Wu, H.; Zhang, C.; Feng, J.; Chen, L.; Xie, R.; Wang, F.; Chen, X.; Zhou, H.; Sun, H.; et al. Clinical, Genetics, and Bioinformatic Characterization of Mutations Affecting an Essential Region of PLS3 in Patients with BMND18. Int. J. Endocrinol. 2018, 2018, 8953217.
- van Dijk, F.S.; Semler, O.; Etich, J.; Köhler, A.; Jimenez-Estrada, J.A.; Bravenboer, N.; Claeys, L.; Riesebos, E.; Gegic, S.; Piersma, S.R.; et al. Interaction between KDELR2 and HSP47 as a Key Determinant in Osteogenesis Imperfecta Caused by Bi-allelic Variants in KDELR2. Am. J. Hum. Genet. 2020, 107, 989–999.
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15.
- Lu, J.T.; Campeau, P.M.; Lee, B.H. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N. Engl. J. Med. 2014, 371, 593–596.
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10.
- Nakamura, Y.; Onodera, S.; Takano, M.; Katakura, A.; Nomura, T.; Azuma, T. Development of a targeted gene panel for the diagnosis of Gorlin syndrome. Int. J. Oral Maxillofac. Surg. 2022, 51, 1431–1444.
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704.
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 2018, 20, 1609–1616.
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360.
- Andersson, K.; Malmgren, B.; Åström, E.; Nordgren, A.; Taylan, F.; Dahllöf, G. Mutations in COL1A1/A2 and CREB3L1 are associated with oligodontia in osteogenesis imperfecta. Orphanet J. Rare Dis. 2020, 15, 80.
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203.
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680.
- Rojas, A.; Aguilar, R.; Henriquez, B.; Lian, J.B.; Stein, J.L.; Stein, G.S.; van Wijnen, A.J.; van Zundert, B.; Allende, M.L.; Montecino, M. Epigenetic control of the bone-master Runx2 gene during osteoblast-lineage commitment by the histone demethylase JARID1B/KDM5B. J. Biol. Chem. 2015, 290, 28329–28342.
- Yu, W.; Chen, K.; Ye, G.; Wang, S.; Wang, P.; Li, J.; Zheng, G.; Liu, W.; Lin, J.; Su, Z.; et al. SNP-adjacent super enhancer network mediates enhanced osteogenic differentiation of MSCs in ankylosing spondylitis. Hum. Mol. Genet. 2021, 26, 277–293.
- Sun, Y.; Miao, N.; Sun, T. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 2019, 156, 29.
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.K.; Sheffield, N.C.; Gräf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705.
- Liu, Y.; Chang, J.C.; Hon, C.C.; Fukui, N.; Tanaka, N.; Zhang, Z.; Lee, M.T.M.; Minoda, A. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 2018, 8, 15499.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
752
Revisions:
2 times
(View History)
Update Date:
11 Sep 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No