 +1 credit
+1 credit
Video Upload Options
The Corona Virus Disease 2019 (COVID-19) pandemic poses a disruptive impact on public health and the global economy. Fortunately, the development of COVID-19 vaccines based on in vitro transcribed messenger RNA (IVT mRNA) has been a breakthrough in medical history, benefiting billions of people with its high effectiveness, safety profile, and ease of large-scale production. This success is the result of decades of continuous RNA research, which has led to significant improvements in the stability and expression level of IVT mRNA through various approaches such as sequence optimization and improved preparation processes. IVT mRNA sequence optimization has been shown to have a positive effect on enhancing mRNA expression level. The innovation of IVT mRNA purification technology is also indispensable, as the purity of IVT mRNA directly affects success of downstream vaccine preparation processes and the potential for inducing unwanted side effects in therapeutic applications. Despite the progress made, challenges related to IVT mRNA sequence design and purification still require further attention to enhance the quality of IVT mRNA in the future.
1. Introduction
| Identifier | Target | Sponsor | Name | Route of Administration | Status | Phase |
|---|---|---|---|---|---|---|
| NCT05217641 | HIV (Human Immunodeficiency Virus) | National Institute of Allergy and Infectious Diseases National Institutes of Health Department of Health and Human Services |
BG505 MD39.3 BG505 MD39.3 gp151 BG505 MD39.3 gp151 CD4KO |
I.M | Active, not recruiting | Ⅰ |
| NCT05398796 | Nipah Virus | National Institute of Allergy and Infectious Diseases Moderna TX, Inc. (Cambridge, MA 02139, USA). National Institutes of Health Clinical Center |
mRNA-1215 | I.M | Recruiting | Ⅰ |
| NCT05430958 | Coronavirus | Inovio Pharmaceuticals | INO-4800 INO-9112 |
I.M | Withdrawn | Ⅰ |
| NCT05414786 | HIV-1 | International AIDS Vaccine Initiative AURUM Tembisa Clinical Research Center for Family Health Research |
mRNA-1644 | I.P | Active, not recruiting | Ⅰ |
| NCT05127434 | Respiratory Syncytial Virus | Moderna TX, Inc. | mRNA-1345 | I.M | Recruiting | Ⅱ/Ⅲ |
| NCT03713086 | Rabies | CureVac | CV7202 | I.M | Completed | Ⅰ |
| NCT05624606 | Influenza Immunization | Sanofi Pasteur | MRT5410 | I.M | Not yet recruiting | Ⅰ/Ⅱ |
| NCT05553301 | Influenza Immunization | Sanofi Pasteur | MRT5407 | I.M | Recruiting | Ⅰ/Ⅱ |
| NCT05105048 | Cytomegalovirus | Moderna TX, Inc. | mRNA-1647 | I.M | Recruiting | Ⅰ |
| NCT05085366 | Cytomegalovirus | Moderna TX, Inc. | mRNA-1647 | I.M | Recruiting | Ⅲ |
| NCT04232280 | Cytomegalovirus | Moderna TX, Inc. | mRNA-1647 | I.M | Active, not recruiting | Ⅱ |
| NCT03382405 | Cytomegalovirus | Moderna TX, Inc. | mRNA-1647/ mRNA-1443 |
I.M | Completed | Ⅰ |
| NCT05164094 | Epstein–Barr Virus | Moderna TX, Inc. | mRNA-1189 | I.M | Recruiting | Ⅰ |
| NCT03392389 | Human Metapneumovirus and Human Parainfluenza | Moderna TX, Inc. | mRNA-1653 | I.M | Completed | Ⅰ |
| NCT05581641 | Malaria | BioNTech SE | BNT165b1 | I.M | Not yet recruiting | Ⅰ |
| NCT04917861 | Zika Virus | Moderna TX, Inc. | mRNA-1893 | I.M | Active, not recruiting | Ⅱ |
| NCT04064905 | Zika Virus | Moderna TX, Inc. Biomedical Advanced Research and Development Authority | mRNA-1893 | I.M | Completed | Ⅰ |
| NCT03014089 | Zika Virus | Moderna TX, Inc. Biomedical Advanced Research and Development Authority | mRNA-1325 | I.M | Completed | Ⅰ |
| NCT05566639 | Seasonal Influenza | Moderna TX, Inc. | mRNA-1010 | I.M | Recruiting | Ⅲ |
| NCT05537038 | Tuberculosis | BioNTech SE | BNT164a1/BNT164b1 | I.M | Not yet recruiting | Ⅰ |
| NCT02888756 | HIV | Rob Gruters Institut d’Investigacions Biomèdiques August Pi i Sunyer IrsiCaixa |
iHIVARNA-01 Tri Mix |
I.M | Terminated Has Results |
Ⅱ |
| NCT05547464 | Tuberculosis | BioNTech SE | BNT164a1/BNT614b1 | I.M | Not yet recruiting | Ⅰ |
| NCT05415462 | Seasonal Influenza | Moderna TX, Inc. | mRNA-1010 | I.M | Active, not recruiting | Ⅲ |
| NCT04956575 | Seasonal Influenza | Moderna TX, Inc. | mRNA-1010 | I.M | Completed | Ⅰ/Ⅱ |
| NCT05333289 | Seasonal Influenza | Moderna TX, Inc. | mRNA-1030/mRNA-102/mRNA-1010 | I.M | Active, not recruiting | Ⅰ/Ⅱ |
| NCT02241135 | Rabies | CureVac | CV7201 | I.M | Completed | Ⅰ |
| NCT05606965 | Influenza | Moderna TX, Inc. | mRNA-1010 | I.M | Recruiting | Ⅱ |
| NCT05252338 | Seasonal Influenza | CureVac GlaxoSmithKline |
CVSQIV | I.M | Recruiting | Ⅰ |
| NCT03345043 | Influenza A(H7N9) | Moderna TX, Inc. | mRNA-1851 | I.M | Completed | Ⅰ |
| NCT03076385 | Influenza A(H10N8) | Moderna TX, Inc. | mRNA-1440 | I.M | Completed | Ⅱ |
| NCT05220975 | RSV | Moderna TX, Inc. | mRNA-1345 | I.M | Recruiting | Ⅲ |
| NCT04144348 | hMPV/PIV3 | Moderna TX, Inc. | mRNA-1653 | I.M | Recruiting | Ⅲ |
| NCT04062669 | Rabies | GlaxoSmithKline | GSK3903133A | I.M | Active, not recruiting | Ⅰ |
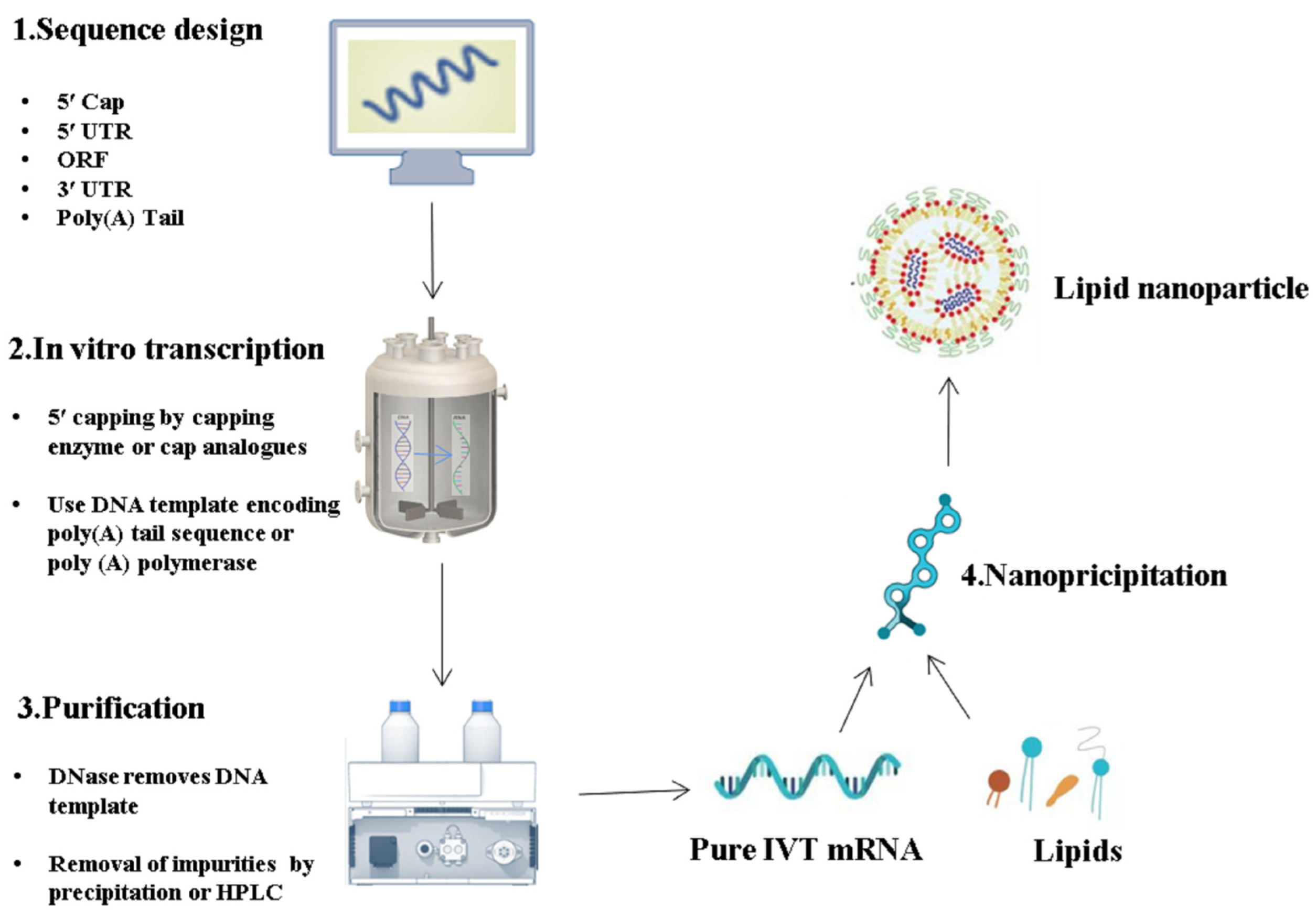
2. Latest Optimization Strategies for IVT mRNA Sequence Design
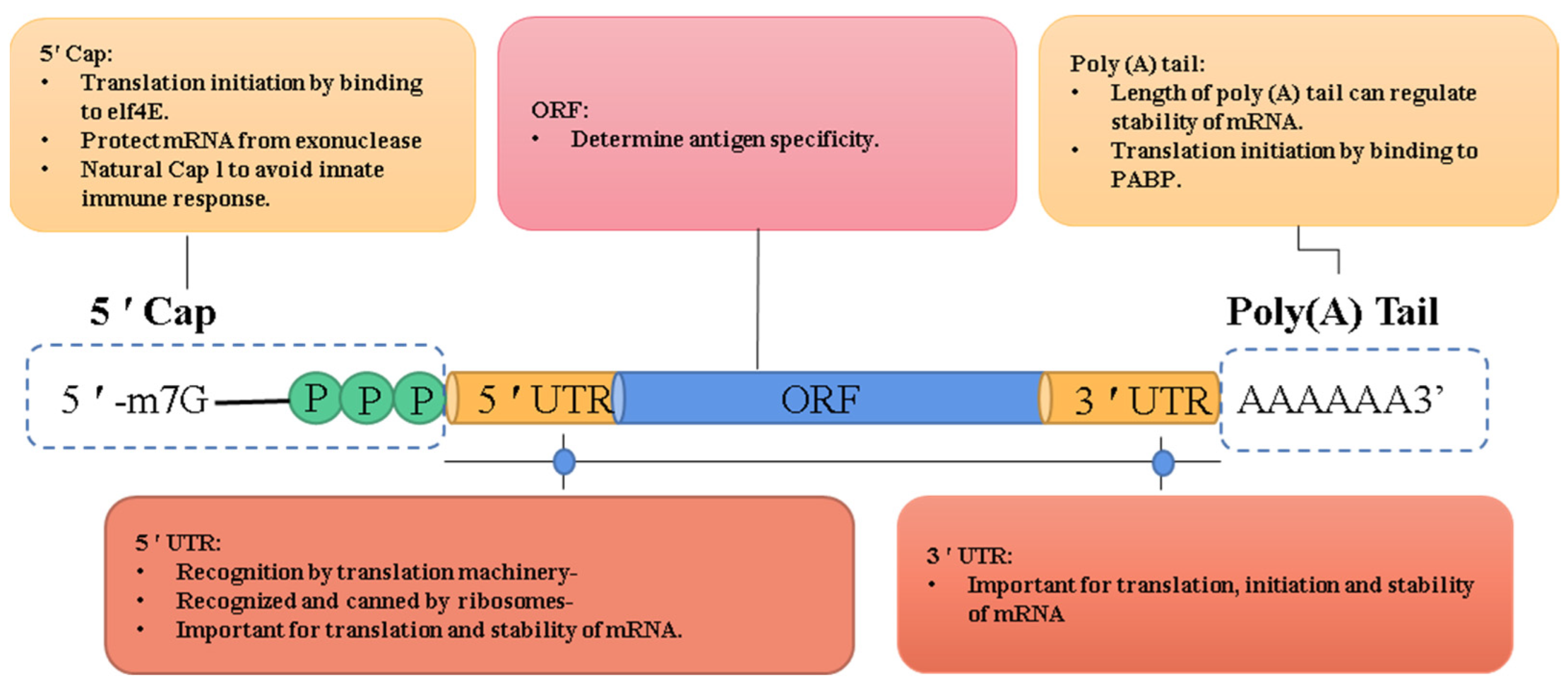
2.1. ORF
2.2. 5′ Cap
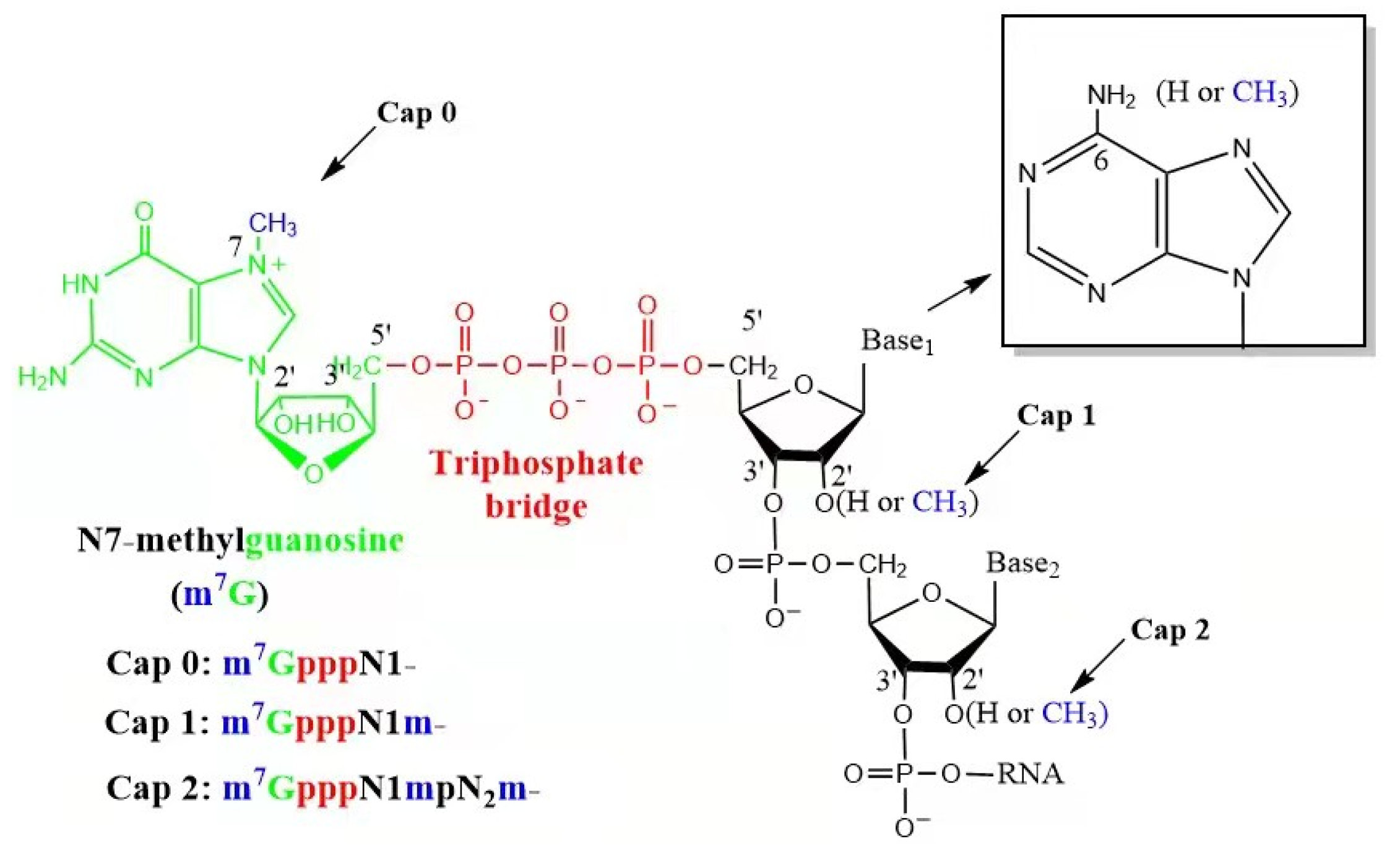

| Enzymatic Capping | CleanCap | |
|---|---|---|
| Enzymes | RNA polymerase; Capping enzyme | RNA polymerase |
| Reaction steps | Transcription and Capping | a one-pot synthesis |
| Purification steps | 2 | 1 |
| Other materials | / | a cap analogue |
| T7 promoter | TAATACGACTCACTATAGGG | TAATACGACTCACTATAAGA |
| Same materials | DNA template; Magnesium-containing buffer; RNase inhibitor, ATP/GTP/CTP/UTP; Inorganic pyrophosphatase | |
2.3. Poly(A) Tail
2.4. UTR
3. IVT mRNA Purification
3.1. The Importance of IVT mRNA Purifications
3.2. Precipitation Methods
3.3. Chromatography Purification Methods
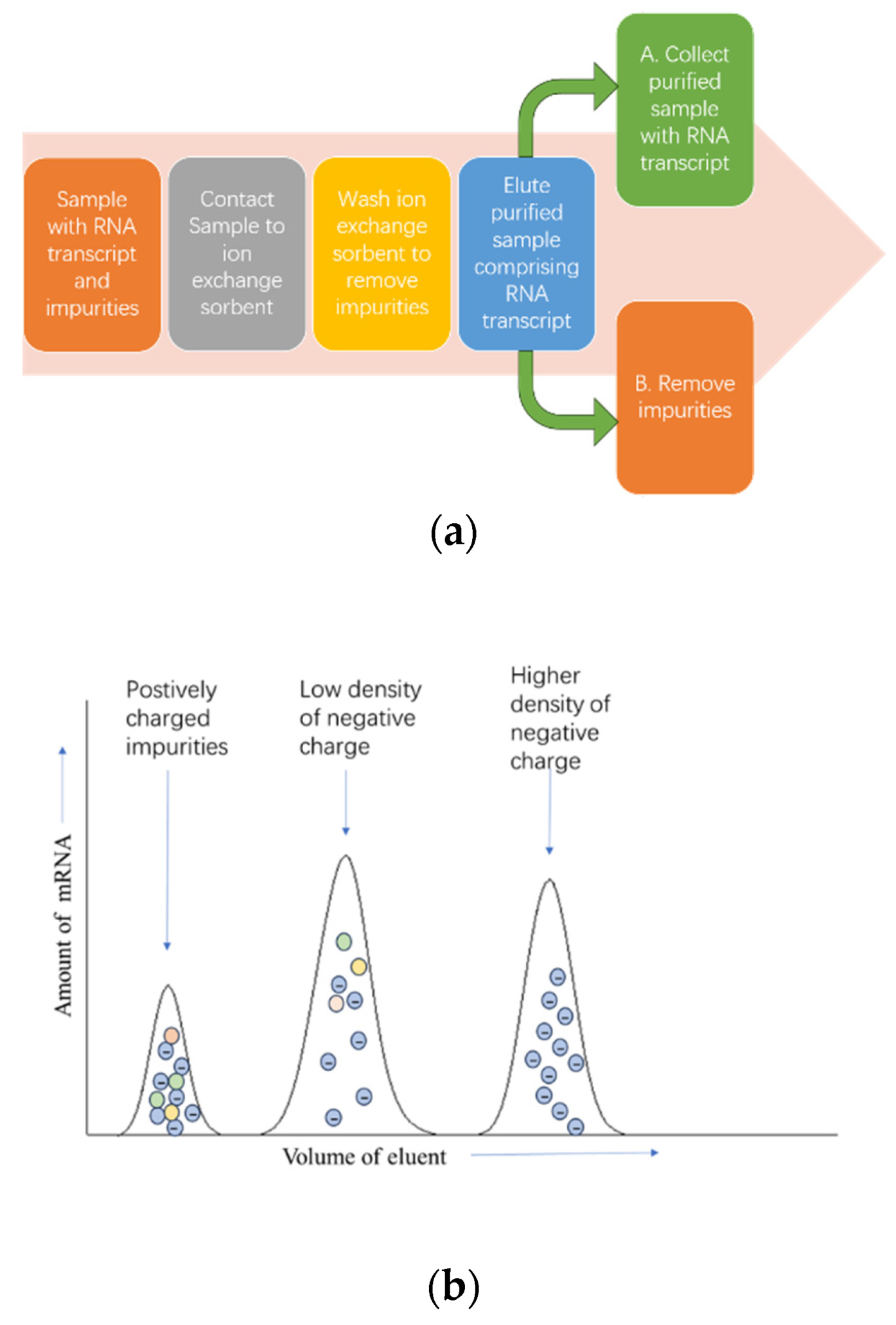
3.4. Non-Chromatography Purification Method
| Methods | Advantages | Disadvantages | |
|---|---|---|---|
| Precipitation method | Precipitation method | easy to operate | form large particles; abnormal mRNA; cationic impurities |
| Non-chromatography purification method | RNase III | effectively remove dsRNA | harm for the secondary structure of mRNA; increases the cost of purification process |
| Lower concentration of Mg2+ | reduce the dsRNA generation | affects the overall yield of the IVT process | |
| Add dispersant into the transcription system | controls the content of dsRNA | / | |
| TFF | fast and efficient | / | |
| Chromatography | SEC | simple | removes unreacted nucleotides, enzymes, short abortion transcripts, and high-molecular-weight DNA templates; time-consuming; difficult to remove impurities of similar size |
| IEC | scalable and cost-effective | / | |
| RP-HPLC | effectively removes dsRNA | toxic organic solvents; may not be conducive to maintaining the stability and biological activity of the target mRNA; loading capacity of column is limited | |
| Affinity HPLC | simple and reliable | low binding capacities and a less cost-effective process | |
| Cellulose chromatography | for large-scale production of IVT mRNA | unclear whether this method can distinguish the inherent secondary structure of dsRNA and mRNA | |
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615.
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733.
- Roest, S.; Hoek, R.A.S.; Manintveld, O.C. BNT162b2 mRNA COVID-19 Vaccine in a Nationwide Mass Vaccination Setting. N. Engl. J. Med. 2021, 384, 1968–1970.
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.L.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.; Lutrick, K.; et al. Interim Estimates of Vaccine Effectiveness of BNT162b2 and mRNA-1273 COVID-19 Vaccines in Preventing SARS-CoV-2 Infection among Health Care Personnel, First Responders, and Other Essential and Frontline Workers—Eight U.S. Locations, December 2020–March 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 495–500.
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838.
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931.
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166.
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279.
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923.
- Youn, H.; Chung, J.K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348.
- Kwon, S.; Kwon, M.; Im, S.; Lee, K.; Lee, H. mRNA vaccines: The most recent clinical applications of synthetic mRNA. Arch. Pharm. Res. 2022, 45, 245–262.
- Kis, Z.; Shattock, R.; Shah, N.; Kontoravdi, C. Emerging Technologies for Low-Cost, Rapid Vaccine Manufacture. Biotechnol. J. 2019, 14, 1800376.
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571.
- Crommelin, D.J.A.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of mRNA Vaccine Stability. J. Pharm. Sci. 2021, 110, 997–1001.
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. In mRNA Vaccines; Part of the Current Topics in Microbiology and Immunology Book Series; Springer International Publishing: Cham, Switzerland, 2020.
- Hornung, V.; Barchet, W.; Schlee, M.; Hartmann, G. RNA recognition via TLR7 and TLR8. In Toll-like Receptors (TLRs) and Innate Immunity; Part of the Handbook of Experimental Pharmacology Book Series; Springer: Berlin/Heidelberg, Germany, 2008; pp. 71–86.
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142.
- Akash, M.S.H.; Rehman, K. (Eds.) Column Chromatography. In Essentials of Pharmaceutical Analysis; Springer: Singapore, 2020; pp. 167–174. ISBN 978-981-15-1547-7.
- Hussain, A.; Yang, H.; Zhang, M.; Liu, Q.; Alotaibi, G.; Irfan, M.; He, H.; Chang, J.; Liang, X.J.; Weng, Y.; et al. mRNA vaccines for COVID-19 and diverse diseases. J. Control Release 2022, 345, 314–333.
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20.
- Naik, R.; Peden, K. Regulatory Considerations on the Development of mRNA Vaccines. In mRNA Vaccines; Part of the Current Topics in Microbiology and Immunology Book Series; Springer International Publishing: Cham, Switzerland, 2020.
- Rice, A.M.; Castillo Morales, A.; Ho, A.T.; Mordstein, C.; Muhlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Evidence for Strong Mutation Bias toward, and Selection against, U Content in SARS-CoV-2: Implications for Vaccine Design. Mol. Biol. Evol. 2021, 38, 67–83.
- Sullenger, B.A.; Nair, S. From the RNA world to the clinic. Science 2016, 352, 1417–1420.
- Cannarozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A role for codon order in translation dynamics. Cell 2010, 141, 355–367.
- Shabalina, S.A.; Spiridonov, N.A.; Kashina, A. Sounds of silence: Synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013, 41, 2073–2094.
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30.
- Spencer, P.S.; Siller, E.; Anderson, J.F.; Barral, J.M. Silent substitutions predictably alter translation elongation rates and protein folding efficiencies. J. Mol. Biol. 2012, 422, 328–335.
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542.
- Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65.
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193.
- To, K.K.W.; Cho, W.C.S. An overview of rational design of mRNA-based therapeutics and vaccines. Expert Opin. Drug Discov. 2021, 16, 1307–1317.
- Drazkowska, K.; Tomecki, R.; Warminski, M.; Baran, N.; Cysewski, D.; Depaix, A.; Kasprzyk, R.; Kowalska, J.; Jemielity, J.; Sikorski, P.J. 2’-O-Methylation of the second transcribed nucleotide within the mRNA 5’ cap impacts the protein production level in a cell-specific manner and contributes to RNA immune evasion. Nucleic Acids Res. 2022, 50, 9051–9071.
- Sikorski, P.J.; Warminski, M.; Kubacka, D.; Ratajczak, T.; Nowis, D.; Kowalska, J.; Jemielity, J. The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5’ cap modulates protein expression in living cells. Nucleic Acids Res. 2020, 48, 1607–1626.
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 2017, 541, 371–375.
- Linares-Fernandez, S.; Lacroix, C.; Exposito, J.Y.; Verrier, B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323.
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 2021, 595, 572–577.
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022, 7, 94.
- Urbina, F.; Morales-Pison, S.; Maldonado, E. Enzymatic Protein Biopolymers as a Tool to Synthetize Eukaryotic Messenger Ribonucleic Acid (mRNA) with Uses in Vaccination, Immunotherapy and Nanotechnology. Polymers 2020, 12, 1633.
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 Messenger RNA Synthesis with Co-transcriptional CleanCap® Analog by In Vitro Transcription. Curr. Protoc. 2021, 1, e39.
- Inagaki, M.; Abe, N.; Li, Z.; Nakashima, Y.; Acharyya, S.; Ogawa, K.; Kawaguchi, D.; Hiraoka, H.; Banno, A.; Meng, Z.; et al. Cap analogs with a hydrophobic photocleavable tag enable facile purification of fully capped mRNA with various cap structures. Nat. Commun. 2023, 14, 2657.
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2021, 9, 3, Erratum in Vaccines 2021, 9, 205.
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001.
- Whitelaw, E.; Coates, A.; Proudfoot, N.J. Globin gene transcripts can utilize histone gene 3’ end processing signals. Nucleic Acids Res. 1986, 14, 7059–7070.
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Tureci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017.
- Goss, D.J.; Kleiman, F.E. Poly(A) binding proteins: Are they all created equal? Wiley Interdiscip. Rev. RNA 2013, 4, 167–179.
- Yu, S.; Kim, V.N. A tale of non-canonical tails: Gene regulation by post-transcriptional RNA tailing. Nat. Rev. Mol. Cell Biol. 2020, 21, 542–556.
- Tang, T.T.L.; Passmore, L.A. Recognition of Poly(A) RNA through Its Intrinsic Helical Structure. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 21–30.
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157.
- Grier, A.E.; Burleigh, S.; Sahni, J.; Clough, C.A.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. pEVL: A Linear Plasmid for Generating mRNA IVT Templates with Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306.
- Lima, S.A.; Chipman, L.B.; Nicholson, A.L.; Chen, Y.H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063.
- Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines 2015, 14, 265–281.
- Trepotec, Z.; Geiger, J.; Plank, C.; Aneja, M.K.; Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 2019, 25, 507–518.
- Preiss, T.; Muckenthaler, M.; Hentze, M.W.J.R. Poly(A)-tail-promoted translation in yeast: Implications for translational control. RNA 1998, 4, 1321–1331.
- Stadler, C.R.; Bahr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Kariko, K.; Tureci, O.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817.
- Xia, X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines 2021, 9, 734.
- Zarghampoor, F.; Azarpira, N.; Khatami, S.R.; Behzad-Behbahani, A.; Foroughmand, A.M. Improved translation efficiency of therapeutic mRNA. Gene 2019, 707, 231–238.
- Balzer Le, S.; Onsager, I.; Lorentzen, J.A.; Lale, R. Dual UTR-A novel 5’ untranslated region design for synthetic biology applications. Synth. Biol. 2020, 5, ysaa006.
- Leppek, K.; Das, R.; Barna, M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174.
- El Mouali, Y.; Balsalobre, C. 3’untranslated regions: Regulation at the end of the road. Curr. Genet. 2019, 65, 127–131.
- Mayr, C. Regulation by 3’-Untranslated Regions. Annu. Rev. Genet. 2017, 51, 171–194.
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150.
- Eberhardt, W.; Doller, A.; Akool, E.S.; Pfeilschifter, J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 2007, 114, 56–73.
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534.
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 2018, 15, 756–762.
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470.
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Lower, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3’ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836.
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809.
- Fukuda, R.; Iwakura, Y.; Ishihama, A. Heterogeneity of RNA polymerase in Escherichia coli. I. A new holoenzyme containing a new sigma factor. J. Mol. Biol. 1974, 83, 353–367.
- Weisman, G.A.; Camden, J.M.; Peterson, T.S.; Ajit, D.; Woods, L.T.; Erb, L. P2 receptors for extracellular nucleotides in the central nervous system: Role of P2X7 and P2Y(2) receptor interactions in neuroinflammation. Mol. Neurobiol. 2012, 46, 96–113.
- Jahn, C.E.; Charkowski, A.O.; Willis, D.K. Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J. Microbiol. Methods 2008, 75, 318–324.
- Fleige, S.; Pfaffl, M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Asp. Med. 2006, 27, 126–139.
- Pereira, M.J.; Behera, V.; Walter, N.G. Nondenaturing purification of co-transcriptionally folded RNA avoids common folding heterogeneity. PLoS ONE 2010, 5, e12953.
- Baiersdorfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Kariko, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35.
- Vomelova, I.; Vanickova, Z.; Sedo, A. Methods of RNA purification. All ways (should) lead to Rome. Folia Biol. 2009, 55, 243–251.
- Green, M.R.; Sambrook, J. Precipitation of RNA with Ethanol. Cold Spring Harb. Protoc. 2020, 2020, 101717.
- Walker, S.E.; Lorsch, J. RNA purification--precipitation methods. Methods Enzymol. 2013, 530, 337–343.
- Lukavsky, P.J.; Puglisi, J.D. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA 2004, 10, 889–893.
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251.
- Kim, I.; McKenna, S.A.; Viani Puglisi, E.; Puglisi, J.D. Rapid purification of RNAs using fast performance liquid chromatography (FPLC). RNA 2007, 13, 289–294.
- McKenna, S.A.; Kim, I.; Puglisi, E.V.; Lindhout, D.A.; Aitken, C.E.; Marshall, R.A.; Puglisi, J.D. Purification and characterization of transcribed RNAs using gel filtration chromatography. Nat. Protoc. 2007, 2, 3270–3277.
- Tuttolomondo, M.; Hansen, P.L.; Mollenhauer, J.; Ditzel, H.J. One-step FPLC-size-exclusion chromatography procedure for purification of rDMBT1 6 kb with increased biological activity. Anal. Biochem. 2018, 542, 16–19.
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Tureci, O.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69.
- Easton, L.E.; Shibata, Y.; Lukavsky, P.J. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA 2010, 16, 647–653.
- Wu, M.Z.; Asahara, H.; Tzertzinis, G.; Roy, B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA 2020, 26, 345–360.
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304.
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249.
- Nina Mencin, A.K.; Ličen, J.; Peljhan, S.; Vidič, J.; Černigoj, U.; Kostelec, T.; Štrancar, A.; Sekirnik, R. Increasing Dynamic Binding Capacity of Oligo(dT) for mRNA Purification. BioProcess Int. 2022, 20, 44–47.
- Baronti, L.; Karlsson, H.; Marusic, M.; Petzold, K. A guide to large-scale RNA sample preparation. Anal. Bioanal. Chem. 2018, 410, 3239–3252.
- Azarani, A.; Hecker, K.H. RNA analysis by ion-pair reversed-phase high performance liquid chromatography. Nucleic Acids Res. 2001, 29, E7.
- Aviv, H.; Leder, P. Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc. Natl. Acad. Sci. USA 1972, 69, 1408–1412.
- Korenč, M.; Mencin, N.; Puc, J.; Skok, J.; Šprinzar Nemec, K.; Martinčič Celjar, A.; Gagnon, P.; Štrancar, A.; Sekirnik, R. Chromatographic purification with CIMmultus™ Oligo dT increases mRNA stability mRNA. Cell and Gene Ther. Insights 2021, 9, 1207–1216.
- Jacobsen, N.; Nielsen, P.S.; Jeffares, D.C.; Eriksen, J.; Ohlsson, H.; Arctander, P.; Kauppinen, S. Direct isolation of poly(A)+ RNA from 4 M guanidine thiocyanate-lysed cell extracts using locked nucleic acid-oligo(T) capture. Nucleic Acids Res. 2004, 32, e64.
- Green, M.R.; Sambrook, J. Isolation of Poly(A)(+) Messenger RNA Using Magnetic Oligo(dT) Beads. Cold Spring Harb. Protoc. 2019, 711–714.
- Zhong, Z.; McCafferty, S.; Opsomer, L.; Wang, H.; Huysmans, H.; De Temmerman, J.; Lienenklaus, S.; Portela Catani, J.P.; Combes, F.; Sanders, N.N. Corticosteroids and cellulose purification improve, respectively, the in vivo translation and vaccination efficacy of sa-mRNAs. Mol. Ther. 2021, 29, 1370–1381.
- Foster, J.B.; Choudhari, N.; Perazzelli, J.; Storm, J.; Hofmann, T.J.; Jain, P.; Storm, P.B.; Pardi, N.; Weissman, D.; Waanders, A.J.; et al. Purification of mRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Human Gene Ther. 2019, 30, 168–178.
- Piao, X.; Yadav, V.; Wang, E.; Chang, W.; Tau, L.; Lindenmuth, B.E.; Wang, S.X. Double-stranded RNA reduction by chaotropic agents during in vitro transcription of messenger RNA. Mol. Ther. Nucleic Acids 2022, 29, 618–624.
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200.
- Ouranidis, A.; Davidopoulou, C.; Tashi, R.K.; Kachrimanis, K. Pharma 4.0 Continuous mRNA Drug Products Manufacturing. Pharmaceutics 2021, 13, 1371.
- Zhang, N.N.; Li, X.F.; Deng, Y.Q.; Zhao, H.; Huang, Y.J.; Yang, G.; Huang, W.J.; Gao, P.; Zhou, C.; Zhang, R.R.; et al. A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283.e1216.


