 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nicole Lintern | -- | 3693 | 2023-08-31 10:50:56 | | | |
| 2 | Fanny Huang | Meta information modification | 3693 | 2023-09-01 08:36:03 | | |
Video Upload Options
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest solid malignancies, with a five-year survival of less than 10%. The resistance of the disease and the associated lack of therapeutic response is attributed primarily to its dense, fibrotic stroma, which acts as a barrier to drug perfusion and permits tumour survival and invasion. As clinical trials of chemotherapy (CT), radiotherapy (RT), and targeted agents have not been successful, improving the survival rate in unresectable PDAC remains an urgent clinical need. Photodynamic stromal depletion (PSD) is a recent approach that uses visible or near-infrared light to destroy the desmoplastic tissue. Preclinical evidence suggests this can resensitise tumour cells to subsequent therapies whilst averting the tumorigenic effects of tumour–stromal cell interactions. So far, the pre-clinical studies have suggested that photodynamic therapy (PDT) can successfully mediate the destruction of various stromal elements without increasing the aggressiveness of the tumour.
1. Introduction
2. The PDAC Tumour Microenvironment and Its Regulation
3. PDT Mechanism and Clinical Progress
Clinical Application of PDT for PDAC
4. Role of PDAC Stroma and Implications for PDT

References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160.
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338.
- Schawkat, K.; Manning, M.A.; Glickman, J.N.; Mortele, K.J. Pancreatic Ductal Adenocarcinoma and Its Variants: Pearls and Perils. Radiographics 2020, 40, 1219–1239.
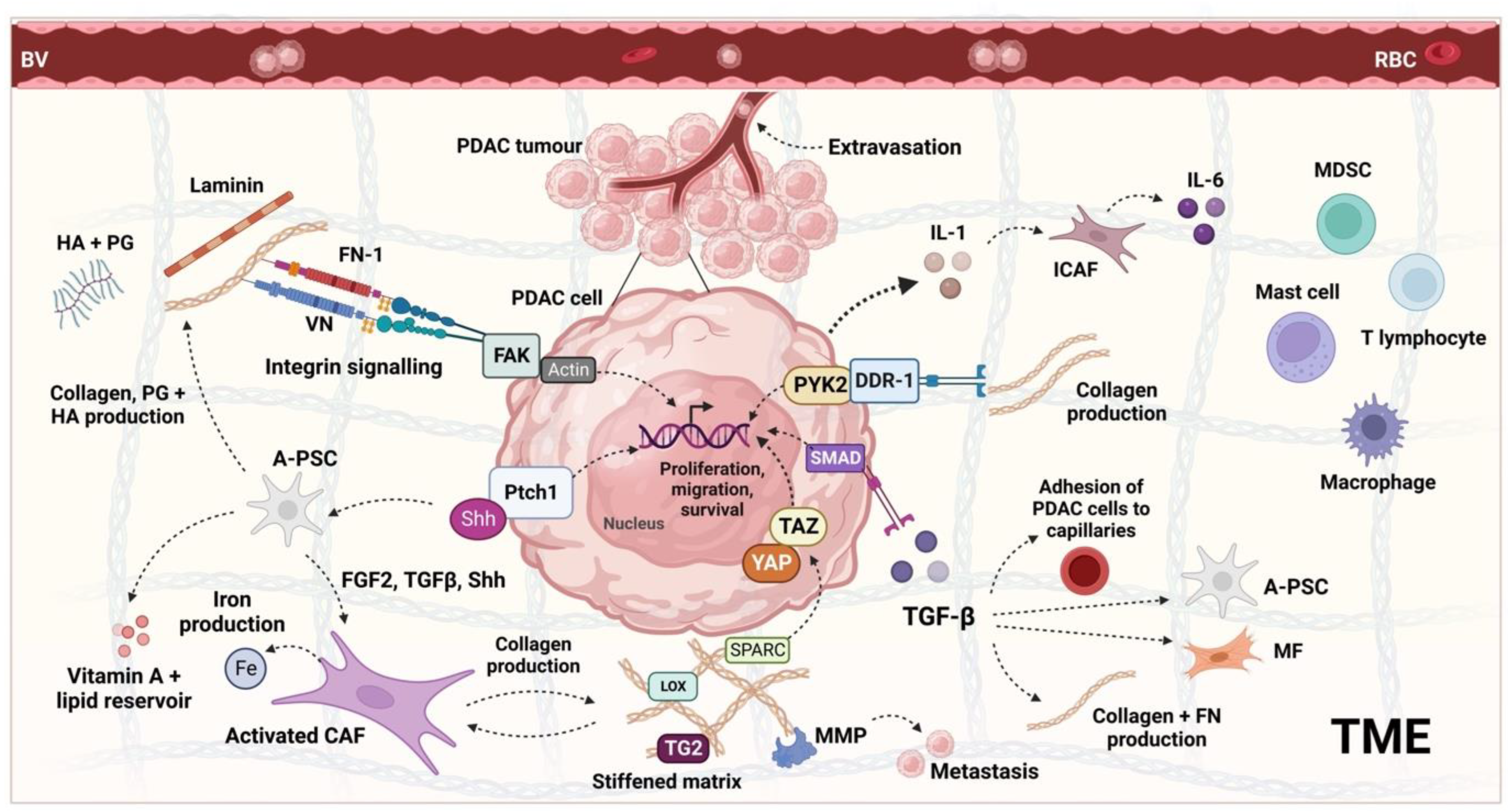
- Ansari, D.; Carvajo, M.; Bauden, M.; Andersson, R. Pancreatic cancer stroma: Controversies and current insights. Scand. J. Gastroenterol. 2017, 52, 641–646.
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271.
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14.
- Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510.
- Du, J.; Gu, J.; Li, J. Mechanisms of drug resistance of pancreatic ductal adenocarcinoma at different levels. Biosci. Rep. 2020, 40, BSR20200401.
- Braun, R.; Klinkhammer-Schalke, M.; Zeissig, S.R.; van Tol, K.K.; Bolm, L.; Honselmann, K.C.; Petrova, E.; Lapshyn, H.; Deichmann, S.; Abdalla, T.S.A.; et al. Clinical Outcome and Prognostic Factors of Pancreatic Adenosquamous Carcinoma Compared to Ductal Adenocarcinoma—Results from the German Cancer Registry Group. Cancers 2022, 14, 3946.
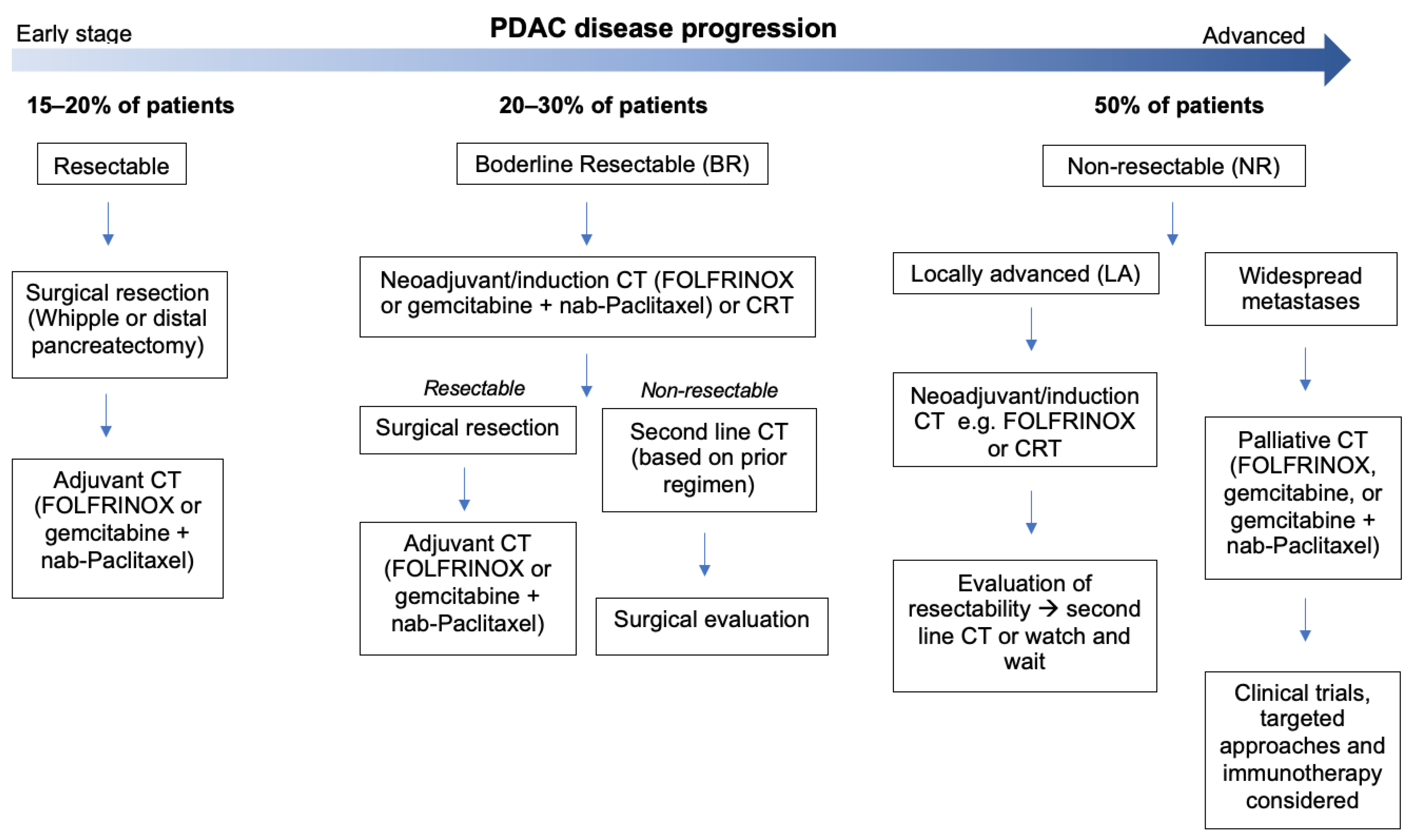
- Evans, D.B.; Ben George, B.; Tsai, S. Non-metastatic Pancreatic Cancer: Resectable, Borderline Resectable, and Locally Advanced-Definitions of Increasing Importance for the Optimal Delivery of Multimodality Therapy. Ann. Surg. Oncol. 2015, 22, 3409–3413.
- Varadhachary, G.R.; Tamm, E.P.; Crane, C.; Evans, D.B.; Wolff, R.A. Borderline resectable pancreatic cancer. Curr. Treat. Opt. Gastroenterol. 2005, 8, 377–384.
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Castillo, C.F.-D.; Hackert, T.; Hayasaki, A.; Katz, M.H.; Kim, S.-W.; Kishiwada, M.; et al. International consensus on definition and criteria of borderline resectable pancreatic ductal adenocarcinoma 2017. Pancreatology 2018, 18, 2–11.
- Nießen, A.; Hackert, T. State-of-the-art surgery for pancreatic cancer. Langenbeck’s Arch. Surg. 2021, 407, 443–450.
- Groot, V.P.; Rezaee, N.; Wu, W.; Cameron, J.L.; Fishman, E.K.; Hruban, R.H.; Weiss, M.J.; Zheng, L.; Wolfgang, C.L.; He, J. Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2018, 267, 936–945.
- Groot, V.P.; Gemenetzis, G.; Blair, A.B.; Rivero-Soto, R.J.; Yu, J.; Javed, A.A.; Burkhart, R.A.; Rinkes, I.H.M.B.; Molenaar, I.Q.; Cameron, J.L.M.; et al. Defining and Predicting Early Recurrence in 957 Patients with Resected Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2019, 269, 1154–1162.
- Parikh, A.A.; Maiga, A.; Bentrem, D.; Squires, M.H., III; Kooby, D.A.; Maithel, S.K.; Weber, S.M.; Cho, C.S.; Katz, M.; Martin, R.C.; et al. Adjuvant Therapy in Pancreas Cancer: Does It Influence Patterns of Recurrence? J. Am. Coll. Surg. 2016, 222, 448–456.
- Shinde, R.S.; Bhandare, M.; Chaudhari, V.; Shrikhande, S.V. Cutting-edge strategies for borderline resectable pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 368–372.
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant Chemotherapy with Gemcitabine and Long-term Outcomes among Patients with Resected Pancreatic Cancer. JAMA 2013, 310, 1473–1481.
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy with Gemcitabine vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267–277.
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406.
- Wu, W.; He, J.; Cameron, J.L.; Makary, M.; Soares, K.; Ahuja, N.; Rezaee, N.; Herman, J.; Zheng, L.; Laheru, D.; et al. The Impact of Postoperative Complications on the Administration of Adjuvant Therapy Following Pancreaticoduode-nectomy for Adenocarcinoma. Ann. Surg. Oncol. 2014, 21, 2873–2881.
- Merkow, R.P.; Bilimoria, K.Y.; Tomlinson, J.S.; Paruch, J.L.; Fleming, J.B.; Talamonti, M.S.; Ko, C.Y.; Bentrem, D.J. Postoperative Complications Reduce Adjuvant Chemotherapy Use in Resectable Pancreatic Cancer. Ann. Surg. 2014, 260, 372–377.
- Åkerberg, D.; Björnsson, B.; Ansari, D. Factors influencing receipt of adjuvant chemotherapy after surgery for pancreatic cancer: A two-center retrospective cohort study. Scand. J. Gastroenterol. 2016, 52, 56–60.
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjørnbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2015, 55, 265–277.
- Tzeng, C.-W.D.; Cao, H.S.T.; Lee, J.E.; Pisters, P.W.; Varadhachary, G.R.; Wolff, R.A.; Abbruzzese, J.L.; Crane, C.H.; Evans, D.B.; Wang, H.; et al. Treatment Sequencing for Resectable Pancreatic Cancer: Influence of Early Metastases and Surgical Complications on Multimodality Therapy Completion and Survival. J. Gastrointest. Surg. 2013, 18, 16–25.
- Muhammadzai, J.; Haider, K.; Moser, M.; Chalchal, H.; Shaw, J.; Gardiner, D.; Dueck, D.-A.; Ahmed, O.; Brunet, B.; Iqbal, M.; et al. Early discontinuation of adjuvant chemotherapy in patients with early-stage pancreatic cancer correlates with inferior survival: A multicenter population-based cohort study. PLoS ONE 2022, 17, e0263250.
- Schwarz, L.; Bruno, M.; Parker, N.H.; Prakash, L.; Mise, Y.; Lee, J.E.; Vauthey, J.-N.; Aloia, T.A.; Conrad, C.; Fleming, J.B.; et al. Active Surveillance for Adverse Events within 90 Days: The Standard for Reporting Surgical Outcomes after Pancreatectomy. Ann. Surg. Oncol. 2015, 22, 3522–3529.
- Polani, F.; Grierson, P.M.; Lim, K.-H. Stroma-targeting strategies in pancreatic cancer: Past lessons, challenges and prospects. World J. Gastroenterol. 2021, 27, 2105–2121.
- Picozzi, V.J.; Stephen, O.-H.; Edwards, A.; Mandelson, M.T.; Dorer, R.; Rocha, F.G.; Alseidi, A.; Biehl, T.; Traverso, W.L.; Helton, W.S.; et al. Five-Year Actual Overall Survival in Resected Pancreatic Cancer: A Contemporary Single-Institution Experience from a Multidisciplinary Perspective. Ann. Surg. Oncol. 2017, 24, 1722–1730.
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant Chemotherapy with Fluorouracil Plus Folinic Acid vs Gemcitabine Following Pancreatic Cancer Resection. JAMA 2010, 304, 1073–1081.
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy with Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients after R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337.
- Chikhladze, S.; Lederer, A.-K.; Kousoulas, L.; Reinmuth, M.; Sick, O.; Fichtner-Feigl, S.; Wittel, U.A. Adjuvant chemotherapy after surgery for pancreatic ductal adenocarcinoma: Retrospective real-life data. World J. Surg. Oncol. 2019, 17, 185.
- Strobel, O.; Lorenz, P.; Hinz, U.M.; Gaida, M.; König, A.-K.; Hank, T.; Niesen, W.; Kaiser, J.; Al-Saeedi, M.; Bergmann, F.; et al. Actual Five-year Survival after Upfront Resection for Pancreatic Ductal Adenocarcinoma. Ann. Surg. 2020, 275, 962–971.
- Moaven, O.; Clark, C.J.; Russell, G.B.M.; Votanopoulos, K.I.; Howerton, R.; Levine, E.A.; Shen, P. Optimal Adjuvant Treatment Approach after Upfront Resection of Pancreatic Cancer. Ann. Surg. 2020, 274, 1058–1066.
- Lim, K.H.; Chung, E.; Khan, A.; Cao, D.; Linehan, D.; Ben-Josef, E.; Wang-Gillam, A. Neoadjuvant Therapy of Pancreatic Cancer: The Emerging Paradigm? Oncologist 2012, 17, 192–200.
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26.
- Reni, M.; Balzano, G.; Zanon, S.; Zerbi, A.; Rimassa, L.; Castoldi, R.; Pinelli, D.; Mosconi, S.; Doglioni, C.; Chiaravalli, M.; et al. Safety and efficacy of preoperative or postoperative chemotherapy for resectable pancreatic adenocarcinoma (PACT-15): A randomised, open-label, phase 2–3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 413–423.
- Heinrich, S.; Schäfer, M.; Weber, A.; Hany, T.F.; Bhure, U.; Pestalozzi, B.C.; Clavien, P.-A.M. Neoadjuvant Chemotherapy Generates a Significant Tumor Response in Resectable Pancreatic Cancer without Increasing Morbidity. Ann. Surg. 2008, 248, 1014–1022.
- Shridhar, R.; Takahashi, C.; Huston, J.; Meredith, K.L. Neoadjuvant therapy and pancreatic cancer: A national cancer database analysis. J. Gastrointest. Oncol. 2019, 10, 663–673.
- Deng, A.; Wang, C.; Cohen, S.J.; Winter, J.M.; Posey, J.; Yeo, C.; Mallick, A.B. Multi-agent neoadjuvant chemotherapy improves survival in early-stage pancreatic cancer: A National Cancer Database analysis. Eur. J. Cancer 2021, 147, 17–28.
- Deig, C.R.; Sutton, T.L.; Beneville, B.; Trone, K.; Stratton, A.; Gunesch, A.N.; Liu, A.I.; Alrohaibani, A.; Mohebnasab, M.; Bassale, S.; et al. Neoadjuvant Therapy Is Associated with Improved Chemotherapy Delivery and Overall Survival Compared to Upfront Resection in Pancreatic Cancer without Increasing Perioperative Complications. Cancers 2022, 14, 609.
- Kamarajah, S.K.; White, S.A.; Naffouje, S.A.; Salti, G.I.; Dahdaleh, F. Adjuvant Chemotherapy Associated with Survival Benefit Following Neoadjuvant Chemotherapy and Pancreatectomy for Pancreatic Ductal Adenocarcinoma: A Population-Based Cohort Study. Ann. Surg. Oncol. 2021, 28, 6790–6802.
- Brunner, T.B.; Scott-Brown, M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat. Oncol. 2010, 5, 64.
- Mukherjee, S.; Hurt, C.N.; Bridgewater, J.; Falk, S.; Cummins, S.; Wasan, H.; Crosby, T.; Jephcott, C.; Roy, R.; Radhakrishna, G.; et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): A multicentre, randomised, phase 2 trial. Lancet Oncol. 2013, 14, 317–326.
- Wang, D.; Liu, C.; Zhou, Y.; Yan, T.; Li, C.; Yang, Q.; Xu, Y.; Zhao, L.; Pei, Q.; Tan, F.; et al. Effect of neoadjuvant radiotherapy on survival of non-metastatic pancreatic ductal adenocarcinoma: A SEER database analysis. Radiat. Oncol. 2020, 15, 107.
- Wang, D.; Ge, H.; Tian, M.; Li, C.; Zhao, L.; Pei, Q.; Tan, F.; Li, Y.; Ling, C.; Güngör, C. The Survival Effect of Radiotherapy on Stage IIB/III Pancreatic Cancer Undergone Surgery in Different Age and Tumor Site Groups: A Propensity Scores Matching Analysis Based on SEER Database. Front. Oncol. 2022, 12, 799930.
- Saadat, L.V.; Chou, J.F.; Gonen, M.; Soares, K.C.; Kingham, T.P.; Varghese, A.M.; Jarnagin, W.R.; D’angelica, M.I.; Drebin, J.A.; O’reilly, E.M.; et al. Treatment patterns and survival in patients with early-onset pancreatic cancer. Cancer 2021, 127, 3566–3578.
- Ansari, D.; Althini, C.; Ohlsson, H.; Andersson, R. Early-onset pancreatic cancer: A population-based study using the SEER registry. Langenbeck’s Arch. Surg. 2019, 404, 565–571.
- Winer, L.K.; Dhar, V.K.; Wima, K.; Morris, M.C.; Lee, T.C.; Shah, S.A.; Ahmad, S.A.; Patel, S.H. The Impact of Tumor Location on Resection and Survival for Pancreatic Ductal Adenocarcinoma. J. Surg. Res. 2019, 239, 60–66.
- Takeda, T.; Sasaki, T.; Inoue, Y.; Mie, T.; Furukawa, T.; Kanata, R.; Kasuga, A.; Matsuyama, M.; Ozaka, M.; Takahashi, Y.; et al. Comprehensive comparison of clinicopathological characteristics, treatment, and prognosis of borderline resectable pancreatic cancer according to tumor location. Pancreatology 2020, 20, 1123–1130.
- Peixoto, R.D.; Speers, C.; McGahan, C.E.; Renouf, D.J.; Schaeffer, D.F.; Kennecke, H.F. Prognostic factors and sites of metastasis in unresectable locally advanced pancreatic cancer. Cancer Med. 2015, 4, 1171–1177.
- Truty, M.J.; Kendrick, M.L.; Nagorney, D.M.; Smoot, R.L.; Cleary, S.P.; Graham, R.P.; Goenka, A.H.; Hallemeier, C.L.; Haddock, M.G.; Harmsen, W.S.; et al. Factors Predicting Response, Perioperative Outcomes, and Survival Following Total Neoadjuvant Therapy for Borderline/Locally Advanced Pancreatic Cancer. Ann. Surg. 2019, 273, 341–349.
- Miquel, M.; Zhang, S.; Pilarsky, C. Pre-clinical Models of Metastasis in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 748631.
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825.
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703.
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556.
- Thomas, A.G.; Awasthi, N. Targeted therapy for pancreatic cancer: Lessons learned and future opportunities. Dig. Med. Res. 2021, 4, 32.
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286.
- Grinshpun, A.; Zarbiv, Y.; Roszik, J.; Subbiah, V.; Hubert, A. Beyond KRAS: Practical Molecular Targets in Pancreatic Adenocarcinoma. Case Rep. Oncol. 2019, 12, 7–13.
- Tempero, M.; Oh, D.-Y.; Tabernero, J.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.-T.; Starling, N.; Bachet, J.-B.; Chang, H.-M.; et al. Ibrutinib in combination with nab-paclitaxel and gemcitabine for first-line treatment of patients with metastatic pancreatic adenocarcinoma: Phase III RESOLVE study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 600–608.
- Hecht, J.R.; Lonardi, S.; Bendell, J.; Sim, H.-W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Martin, A.J.M.; Park, J.O.; Greil, R.; et al. Randomized Phase III Study of FOLFOX Alone or with Pegilodecakin as Second-Line Therapy in Patients with Metastatic Pancreatic Cancer That Progressed after Gemcitabine (SEQUOIA). J. Clin. Oncol. 2021, 39, 1108–1118.
- Shin, S.; Park, C.M.; Kwon, H.; Lee, K.-H. Erlotinib plus gemcitabine versus gemcitabine for pancreatic cancer: Real-world analysis of Korean national database. BMC Cancer 2016, 16, 443.
- Hammel, P.; Huguet, F.; van Laethem, J.L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients with Locally Advanced Pancreatic Cancer Controlled after 4 Months of Gemcitabine with or without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844–1853.
- Di Marco, M.; Grassi, E.; Durante, S.; Vecchiarelli, S.; Palloni, A.; Macchini, M.; Casadei, R.; Ricci, C.; Panzacchi, R.; Santini, D.; et al. State of the art biological therapies in pancreatic cancer. World J. Gastrointest. Oncol. 2016, 8, 55–66.
- Safran, H.; Miner, T.; Bahary, N.; Whiting, S.; Lopez, C.D.; Sun, W.; Charpentier, K.; Shipley, J.; Anderson, E.; McNulty, B.; et al. Lapatinib and Gemcitabine for Metastatic Pancreatic Cancer. Am. J. Clin. Oncol. 2011, 34, 50–52.
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863.
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 24.
- Jimeno, A.; Weiss, G.J.; Miller, W.H.; Gettinger, S.; Eigl, B.J.; Chang, A.L.S.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I Study of the Hedgehog Pathway Inhibitor IPI-926 in Adult Patients with Solid Tumors. Clin. Cancer Res. 2013, 19, 2766–2774.
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375.
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194.
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin with Two Sides? Front. Oncol. 2020, 10, 576399.
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100.
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br. J. Cancer 2014, 110, 1698–1704.
- DeWitt, J.M.; Sandrasegaran, K.; O’Neil, B.; House, M.G.; Zyromski, N.J.; Sehdev, A.; Perkins, S.M.; Flynn, J.; McCranor, L.; Shahda, S. Phase 1 study of EUS-guided photodynamic therapy for locally advanced pancreatic cancer. Gastrointest. Endosc. 2019, 89, 390–398.
- Hanada, Y.; Pereira, S.P.; Pogue, B.; Maytin, E.V.; Hasan, T.; Linn, B.; Mangels-Dick, T.; Wang, K.K. EUS-guided verteporfin photodynamic therapy for pancreatic cancer. Gastrointest. Endosc. 2021, 94, 179–186.
- Karimnia, V.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodestruction of Stromal Fibroblasts Enhances Tumor Response to PDT in 3D Pancreatic Cancer Coculture Models. Photochem. Photobiol. 2020, 97, 416–426.
- Celli, J.P. Stromal Interactions as Regulators of Tumor Growth and Therapeutic Response: A Potential Target for Photodynamic Therapy? Isr. J. Chem. 2012, 52, 757–766.
- Jafari, R.; Cramer, G.M.; Celli, J.P. Modulation of Extracellular Matrix Rigidity Via Riboflavin-mediated Photocrosslinking Regulates Invasive Motility and Treatment Response in a 3D Pancreatic Tumor Model. Photochem. Photobiol. 2020, 96, 365–372.
- Maneshi, P.; Mason, J.; Dongre, M.; Öhlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485.
- Korc, M. Pancreatic cancer–associated stroma production. Am. J. Surg. 2007, 194, S84–S86.
- Muller, M.; Haghnejad, V.; Schaefer, M.; Gauchotte, G.; Caron, B.; Peyrin-Biroulet, L.; Bronowicki, J.-P.; Neuzillet, C.; Lopez, A. The Immune Landscape of Human Pancreatic Ductal Carcinoma: Key Players, Clinical Implications, and Challenges. Cancers 2022, 14, 995.
- Carstens, J.L.; de Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095.
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700.
- Kpeglo, D.; Hughes, M.D.; Dougan, L.; Haddrick, M.; Knowles, M.A.; Evans, S.D.; Peyman, S.A. Modeling the mechanical stiffness of pancreatic ductal adenocarcinoma. Matrix Biol. Plus 2022, 14, 100109.
- Ogawa, Y.; Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Fujii-Nishimura, Y.; Hori, S.; Yagi, H.; Abe, Y.; Kitago, M.; et al. Three Distinct Stroma Types in Human Pancreatic Cancer Identified by Image Analysis of Fibroblast Subpopulations and Collagen. Clin. Cancer Res. 2021, 27, 107–119.
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- Askan, G.; Sahin, I.H.; Chou, J.F.; Yavas, A.; Capanu, M.; Iacobuzio-Donahue, C.A.; Basturk, O.; O’reilly, E.M. Pancreatic cancer stem cells may define tumor stroma characteristics and recurrence patterns in pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 385.
- Wu, H.; Minamide, T.; Yano, T. Role of photodynamic therapy in the treatment of esophageal cancer. Dig. Endosc. 2019, 31, 508–516.
- Saini, R.; Lee, N.V.; Liu, K.Y.; Poh, C.F. Prospects in the Application of Photodynamic Therapy in Oral Cancer and Premalignant Lesions. Cancers 2016, 8, 83.
- Crous, A.; Abrahamse, H. Photodynamic therapy of lung cancer, where are we? Front. Pharmacol. 2022, 13, 932098.
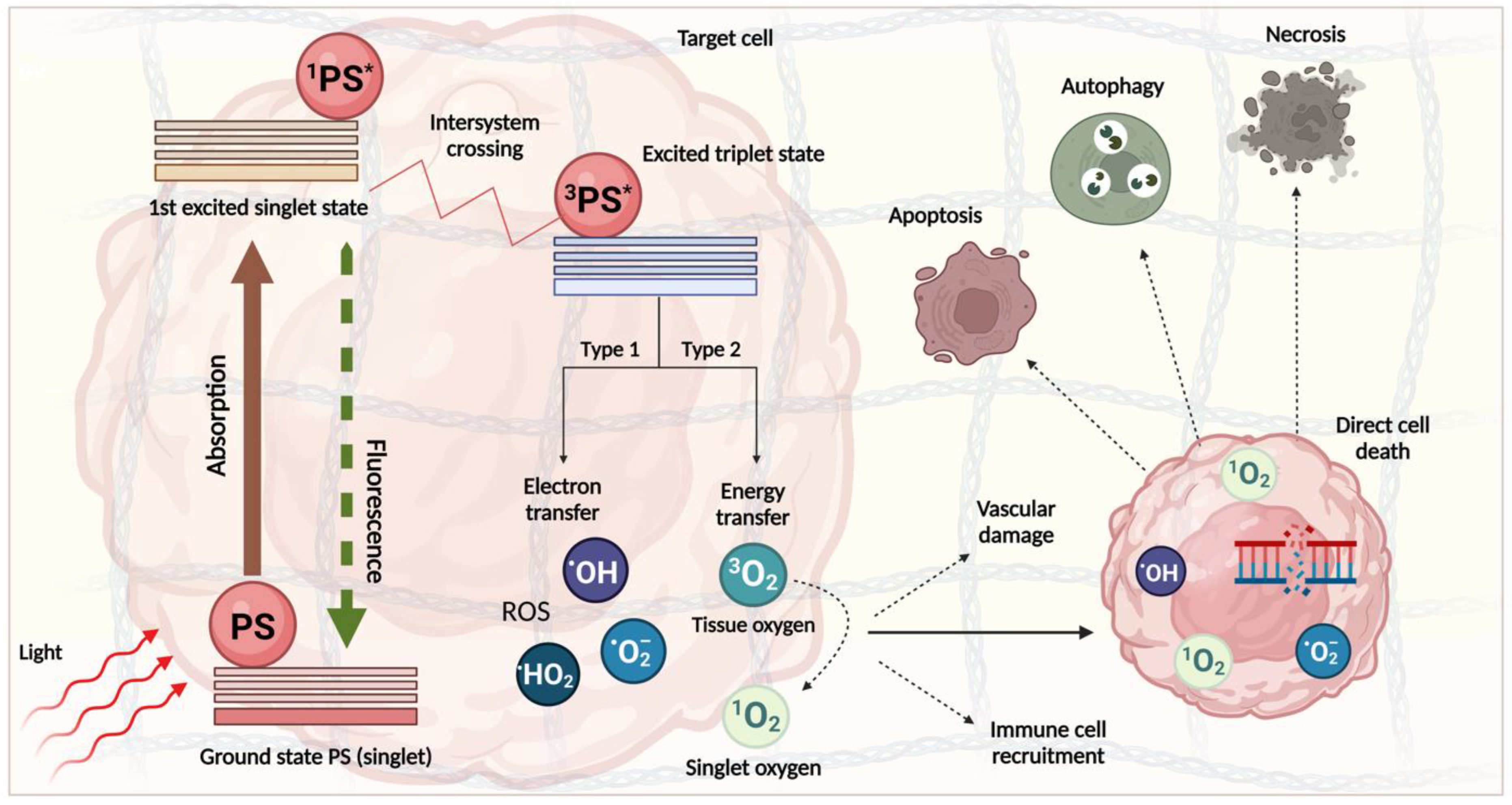
- Robertson, C.A.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B 2009, 96, 1–8.
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in PDT: Part three—Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn. Ther. 2005, 2, 91–106.
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545.
- Mroz, P.; Szokalska, A.; Wu, M.X.; Hamblin, M.R. Photodynamic Therapy of Tumors Can Lead to Development of Systemic Antigen-Specific Immune Response. PLoS ONE 2010, 5, e15194.
- Mashayekhi, V.; Op’t Hoog, C.; Oliveira, S. Vascular targeted photodynamic therapy: A review of the efforts towards molecular targeting of tumor vasculature. J. Porphyr. Phthalocyanines 2019, 23, 1229–1240.
- Abrahamse, H.; Hamblin Michael, R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364.
- Liu, K.-H.; Wang, C.-P.; Chang, M.-F.; Chung, Y.-W.; Lou, P.-J.; Lin, J.-H. Molecular characterization of photosensitizer-mediated photodynamic therapy by gene expression profiling. Hum. Exp. Toxicol. 2013, 33, 629–637.
- Kessel, D. Correlation between subcellular localization and photodynamic efficacy. J. Porphyr. Phthalocyanines 2004, 8, 1009–1014.
- Uchoa, A.F.; Oliveira, C.S.; Baptista, M.S. Relationship between structure and photoactivity of porphyrins derived from pro-toporphyrin IX. J. Porphyr. Phthalocyanines 2010, 14, 832–845.
- Zhen, Z.; Tang, W.; Chuang, Y.-J.; Todd, T.; Zhang, W.; Lin, X.; Niu, G.; Liu, G.; Wang, L.; Pan, Z.; et al. Tumor Vasculature Targeted Photodynamic Therapy for Enhanced Delivery of Nanoparticles. ACS Nano 2014, 8, 6004–6013.
- Bown, S.G.; Rogowska, A.Z.; Whitelaw, D.E.; Lees, W.-R.; Lovat, L.B.; Ripley, P.; Jones, L.; Wyld, P.; Gillams, A.; Hatfield, A.W.R. Photodynamic therapy for cancer of the pancreas. Gut 2002, 50, 549–557.
- Choi, J.-H.; Oh, D.; Lee, J.H.; Park, J.-H.; Kim, K.-P.; Lee, S.S.; Lee, Y.-J.; Lim, Y.-S.; Song, T.J.; Lee, S.S.; et al. Initial human experience of endoscopic ultrasound-guided photodynamic therapy with a novel photosensitizer and a flexible laser-light catheter. Endoscopy 2015, 47, 1035–1038.
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.P.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1, 1.
- Gündel, B.; Liu, X.; Löhr, M.; Heuchel, R. Pancreatic Ductal Adenocarcinoma: Preclinical in vitro and ex vivo Models. Front. Cell Dev. Biol. 2021, 9, 741162.
- Voutouri, C.; Polydorou, C.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Hyaluronan-Derived Swelling of Solid Tumors, the Contribution of Collagen and Cancer Cells, and Implications for Cancer Therapy. Neoplasia 2016, 18, 732–741.
- Boucher, Y.; Jain, R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: Implications for vascular collapse. Cancer Res. 1992, 52, 5110–5114.
- DuFort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Brockenbrough, J.S.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119.
- Zhao, T.; Zhang, R.; He, Q.; Zhou, H.; Song, X.; Gong, T.; Zhang, Z. Partial ligand shielding nanoparticles improve pancreatic ductal adenocarcinoma treatment via a multifunctional paradigm for tumor stroma reprogramming. Acta Biomater. 2022, 145, 122–134.
- Yang, H.; Liu, R.; Xu, Y.; Qian, L.; Dai, Z. Photosensitizer Nanoparticles Boost Photodynamic Therapy for Pancreatic Cancer Treatment. Nano-Micro Lett. 2021, 13, 35.
- Shah, V.; Sheppard, B.; Sears, R.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70.

