Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Rosaria Miranda | -- | 10442 | 2023-08-30 19:35:35 | | | |
| 2 | Camila Xu | Meta information modification | 10442 | 2023-08-31 04:24:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Miranda, M.R.; Vestuto, V.; Moltedo, O.; Manfra, M.; Campiglia, P.; Pepe, G. Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/48661 (accessed on 23 July 2026).
Miranda MR, Vestuto V, Moltedo O, Manfra M, Campiglia P, Pepe G. Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/48661. Accessed July 23, 2026.
Miranda, Maria Rosaria, Vincenzo Vestuto, Ornella Moltedo, Michele Manfra, Pietro Campiglia, Giacomo Pepe. "Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases" Encyclopedia, https://encyclopedia.pub/entry/48661 (accessed July 23, 2026).
Miranda, M.R., Vestuto, V., Moltedo, O., Manfra, M., Campiglia, P., & Pepe, G. (2023, August 30). Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases. In Encyclopedia. https://encyclopedia.pub/entry/48661
Miranda, Maria Rosaria, et al. "Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases." Encyclopedia. Web. 30 August, 2023.
Copy Citation
Ion channels (ICs) are integral membrane proteins that play a crucial role in regulating the ions’ flow across cell membranes. They are essential for maintaining cellular homeostasis and are involved in various physiological processes. The pathogenesis of various gastrointestinal (GI) disorders, including gastritis, ulcers, inflammatory bowel disease (IBD) and cancer, can be linked to oxidative stress. It is known that reactive species carry out a crucial role in the genesis and progression of these pathologies; however, the contribution of ionic channels in their development is still under discussion. The function of ion channels in the gastrointestinal tract influences a variety of cellular processes.
ion channels
oxidative stress
reactive species
antioxidants
1. Introduction
Oxidative stress (OS) occurs due to an imbalance between the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and the endogenous antioxidant defense system. This imbalance leads to cellular injury and subsequent dysfunctions that can result in a wide range of disorders [1][2]. Reactive species represent byproducts of normal cellular metabolism and are normally neutralized by a variety of cellular antioxidants: enzymatic, such as catalase (CAT) and superoxide dismutase (SOD), or non-enzymatic, such as glutathione [3].
Reactive species are generated in response to factors like UV radiation, smoking, alcohol consumption, the use of non-steroidal anti-inflammatory drugs (NSAIDs), infections, ischemia-reperfusion injury, and inflammation (Figure 1) [4][5]. While low to moderate levels of ROS and RNS have positive effects on several physiological functions, such as fighting pathogens, promoting wound healing, and aiding in tissue repair, they also serve as important signaling biomolecules [6][7]. However, excessive production can disrupt the body’s balance and lead to oxidative damage in tissues. For this reason, OS has been implicated in various disease processes, including aging, ischemia-reperfusion injury, hypertension, atherosclerosis, diabetic neuropathies, renal diseases, neurological disorders, inflammatory bowel disease and cancer [8][9].
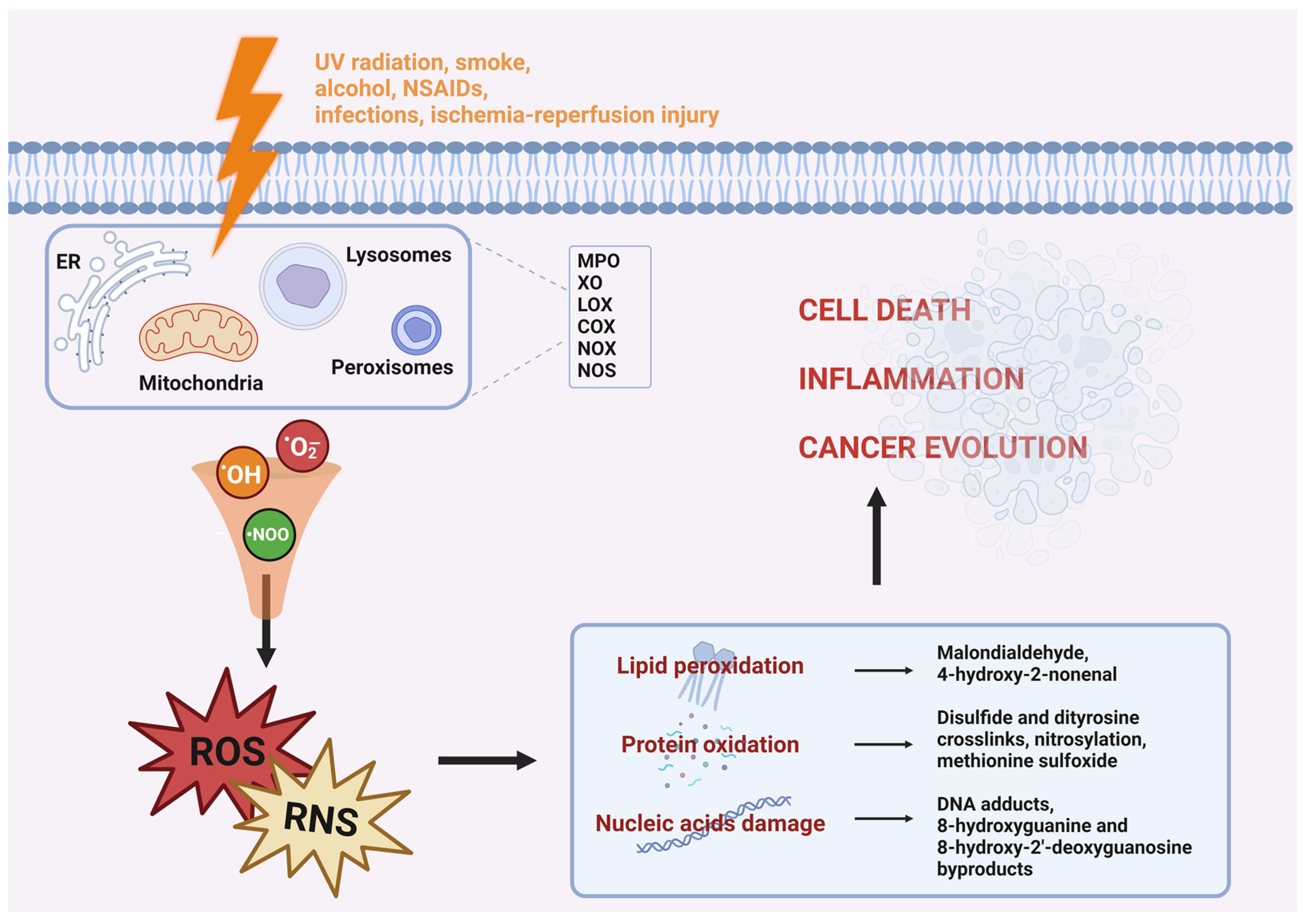
Figure 1. Oxidative stress and its effects on cells. Reactive species are generated by multiple processes at the level of different cell organelles and can be produced in reaction to different noxious stimuli. ROS and RNS thus obtained damaged cellular membranes, proteins, and DNA, contributing to cell death, inflammation and cancer.
In this regard, several exogenous and endogenous bioactive agents are well recognized for their ability to strengthen the antioxidant response against several diseases caused by OS, helping to fight the harmful effects of reactive species and to promote overall health [10][11]. For instance, nutraceuticals represent important sources of different classes of antioxidants and anti-inflammatory compounds, such as polyphenols and many bioactive peptides [12][13], which exhibit remarkable antioxidant activity that can be beneficial in GI diseases [14][15][16] and other conditions related to OS [17][18][19][20]. Furthermore, vitamin E and vitamin C, as well as supplementation with SOD and other endogenous enzymes have been shown to decrease oxidative stress markers and improve disease activity in patients with IBD [21][22].
The GI tract is both a source and a target of OS. Despite the protective barrier provided by the epithelial layer, digested food and pathogens can activate the epithelium, polymorphonuclear neutrophils (PMNs), and macrophages, leading to inflammation and the production of inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6, IL-4 and IL-8) and other mediators involved in oxidative stress [23][24]. Various GI disorders, for instance, gastroduodenal ulcers; GI malignancies, such as gastric and colorectal cancer; gastroesophageal reflux disease (GERD); IBD, which includes Crohn’s disease (CD) and ulcerative colitis (UC); and irritable bowel syndrome (IBS), partly arise due to oxidative stress [7][10]. Considering that OS can alter the normal functioning of ion channels, which are essential for maintaining cellular homeostasis in gastrointestinal cells [25], it is crucial to understand the processes and changes initiated by free radicals.
1.1. Oxidative Stress
OS can have various implications on cellular homeostasis, depending on the intensity and duration of the insult. In some cases, an excess of ROS can damage cellular biomolecules, including lipids, proteins, and nucleic acids, leading to various negative consequences (Figure 1) [26][27].
Damage to lipids leads to the formation of lipid peroxidation products, such as malondialdehyde and 4-hydroxy-2-nonenal, derived from omega-6 fatty acids. These products exhibit various physiological and protective functions as signaling molecules stimulating gene expression and cell survival. However, they can also have a cytotoxic role by inhibiting gene expression and inducing cell death [28].
Lipid peroxidation is triggered by free radicals that affect unsaturated fatty acids in cell membranes. It involves a series of chain reactions continuing to occur once activated, resulting in progressive and cumulative changes in the structure and functions of the membrane [29]. These alterations include abnormalities in membrane potential, mitochondrial fission and its consequent release of calcium, activation of caspase-3, DNA fragmentation and, ultimately, the initiation of programmed cell death [30].
Damage to proteins can affect their structure and function, impairing metabolic pathways and enzymatic activity. Modifications caused by ROS/RNS include the loss of sulfhydryl groups, and the formation of carbonyls, disulfide crosslinks, methionine sulfoxide, dityrosine cross-links, nitration, nitrosylation, glutathionylation and glyoxidation, among others [31][32]. The oxidation of protein sulfhydryl groups occurs as a result of exposure to different ROS and is considered one of the immediate responses to increased oxidative stress. One of the main targets for oxidation within proteins is the amino acid cysteine, which contains a highly reactive thiol group. The latter undergo various oxidative modifications, including the formation of sulfenic, sulfinic, and sulfonic acids depending on the concentration of oxidants, redox potentials, local pH, charge, temperature, etc. [33][34]. Additionally, in pathological conditions with excessively high concentrations of ROS, other amino acids, such as arginine and lysine, can be altered to aldehydes, and methionine residues can be converted to sulfoxides or sulfones. Some of these modifications are irreversible and permanently affect protein function. Another oxidative modification in proteins is the formation of dityrosine crosslinks, which apparently occurs as a result of the reaction between two tyrosyl radicals, generated by peroxidases and other heme proteins. The addition of carbonyl-containing adducts to the side chains of amino acid residues, such as lysine, arginine, proline, and threonine, is another very common post-translational structural alteration in proteins [35].
Damage to nucleic acids by OS includes strand breaks, base modifications, and the formation of DNA adducts. If not adequately repaired, these damages can lead to genomic instability [36][37]. ROS can react with purines and pyrimidines DNA bases causing various byproducts, such as 8-hydroxyguanine and 8-hydroxy-2′-deoxyguanosine, interfering with normal DNA replication and transcription [36]. Consequently, mutations caused by oxidative stress triggers can activate proto-oncogenes, converting them into oncogenes. Conversely, oncosuppressor genes, which normally control cell growth and prevent tumor formation, can be inactivated by ROS-induced DNA damage leading to uncontrolled cell proliferation and the development of gastrointestinal cancers [38][39].
Oxidative stress can also influence cellular signaling regulation and gene expression. It can modulate transcription factors, such as activator protein-1 (AP-1), nuclear factor kappa B (NF-κB), and/or NF-E2-related factor (Nrf2), which control the expression of genes involved in inflammatory response and apoptosis. Additionally, it can interfere with cellular signaling pathways, including mitogen-activated protein kinases (MAPKs) and tyrosine kinases. It can also disrupt the cellular response to various external signals (Figure 1) [40].
1.2. Reactive Oxygen and Nitrogen Species
Molecular oxygen (O2) is involved in energy production through mitochondrial respiration; however, it also paradoxically contributes to cell death because ROS are derived from its chemical reduction. ROS include radical compounds such as superoxide (O2•−), hydroxyl radicals (•OH), lipid hydroperoxides, and non-radical reactive compounds including singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), chloramines (RNHCl) and ozone (O3) [7][41].
Reactive nitrogen species are unstable compounds that include both radical species as nitric oxide (•NO) and nitrogen dioxide (•NO2), and non-radical species as peroxynitrite (ONOO−) and dinitrogen trioxide (N2O3), that possess unpaired electrons in their outermost electron orbit. RNS often interact with reactive oxygen species, such as peroxynitrite, leading to nitrosative stress. RNS are a group of free radicals that are produced by nitric oxide synthase, which is expressed within the intestinal submucosa and mucosal regions [42][43].
ROS and RNS can be generated within various intracellular organelles, including mitochondria, endoplasmic reticulum, peroxisomes, nuclei, cytosol, plasma membranes, and even extracellular spaces (Figure 1). The primary source of ROS in cells is provided by complexes I and III of the mitochondrial electron transport chain, which are responsible for the conversion of approximately 1% to 3% of total oxygen to superoxide. Indeed, it is during cellular respiration that the accumulation of high-energy electrons can result in the production of oxygen-free radicals [44][45].
Moreover, numerous enzymes, such as nicotinamide adenine dinucleotide phosphate oxidase (NOX), phospholipase A2 (PLA2), nitric oxide synthase (NOS), cyclooxygenases (COXs), xanthine oxidase (XO), lipoxygenases (LOXs) and myeloperoxidase (MPO) can generate reactive species as part of their normal metabolic processes (Figure 1) [46].
NADPH oxidase, a membrane-bound enzyme complex present in the plasma membrane and phagosomes of phagocytes, is responsible for respiratory burst that occurs when phagocytic cells consume a large amount of oxygen during phagocytosis, leading to the release of O2•− into the extracellular space or phagosomes. It consists of multiple subunits, including gp91PHOX, p22PHOX, p67PHOX, p47PHOX, p40PHOX, and Rho GTPases. Activation of NADPH oxidase involves the relocation of cytosolic components to the cell membrane, where they assemble and activate the catalytic core, flavocytochrome b. There are six homologs of NADPH oxidase, collectively known as the NOX family, with different intracellular localizations. Among them, NOX1 and DUOX2 (dual oxidase 2) play some significant roles in gastrointestinal pathology, particularly in Helicobacter pylori-induced gastric inflammation, IBD and tumor development. DUOX is found in all segments of the intestine, while NOX1 is present only in the ileum, cecum, and colonic epithelium [7][47][48].
Lipoxygenases are non-heme iron enzymes that catalyze the dioxygenation of polyenoic fatty acids, resulting in the production of hydroperoxyl derivatives such as hydroperoxyeicosatetraenoic acids (HPETEs). HPETEs can be further converted into pro-inflammatory hydroxyl derivatives, leukotrienes and lipoxins. In any case, LOX-mediated oxidation of arachidonic acid generates reactive oxygen species. LOX enzymes are found in various cells of the gastrointestinal tract and play a role in inflammation and diseases such as atherosclerosis, gastric inflammation, and peritonitis [49][50][51].
Myeloperoxidase is a heme-enzyme localized in lysosomes of neutrophils, macrophages, and monocytes. This is an enzyme that generates highly reactive HOCl and other ROS; HOCl reacts with H2O2 to generate single oxygen and chloride ion. MPO activity increases in H. pylori infection and contributes to gastric inflammation and the development of gastric cancer [52].
Nitric oxide synthase is an enzyme that produces the signaling molecule NO. There are three types of NOS: neuronal NOS (nNOS or NOS I), endothelial NOS (eNOS or NOS III), and inducible NOS (iNOS or NOS II) [53]. These isozymes catalyze the conversion of L-arginine to L-citrulline and NO; at low levels of L-arginine, NOS can produce hydrogen peroxide in addition to NO. While NO is a weak oxidant, it can react with superoxide to form peroxynitrite. Additionally, both NO and peroxynitrite in can generate nitrite and nitrate ions, which can accumulate in cells along with their intermediates (e.g., nitrogen dioxide, nitrogen trioxide, nitrogen monoxide). This accumulation can lead to the nitration and nitrosation of nucleic acids, proteins, and lipids, thereby disrupting their function. Nitrated lipids can elicit various physiological responses and can also produce NO [54][55]. NOS is expressed in the GI tract and plays a role in GI mucosal defense and injury. NO is involved in maintaining the normal functions of the GI mucosa and has a cytoprotective role: it regulates gastric mucosal blood flow, epithelial secretion, and barrier function, thereby maintaining GI mucosal integrity. However, excessive expression of iNOS is observed in conditions like chronic ulcerative colitis and peptic ulcers, indicating potential involvement of reactive nitrogen species generated by iNOS in both normal and pathophysiological conditions of the GI tract [7].
Cyclooxygenase is an enzyme that plays a crucial role in the release of arachidonic acid from cell membrane lipids and its subsequent conversion into prostanoids. There are two isoforms of COX: COX-1, constitutively expressed in most tissues and is involved in various physiological functions. COX-2 is an inducible isoform that is constitutively expressed at low levels in some tissues and upregulated in others in response to inflammation and tumorigenesis; and a reported splice variant known as COX-3. The peroxidase activity of COX leads to the generation of radicals, like NAD• and NADP•, which can eventually produce superoxide. Both COX-1 and COX-2 are physiologically found in the gastric mucosa, and enhanced levels are often observed at the edges of ulcers. COX-2 has been linked to early cancerous alterations in the gastrointestinal mucosa, such as Barrett’s esophagus, gastritis induced by H. pylori, and inflamed colonic mucosa. It is also involved in the progression of cancers related to these conditions [56][57].
1.3. Antioxidant Defense in the GI Tract
The human body exhibits two fundamental forms of antioxidant defense: enzymatic and non-enzymatic. The major enzymatic antioxidants include catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPX) and glutathione reductase (GSR) (Figure 2) [4].
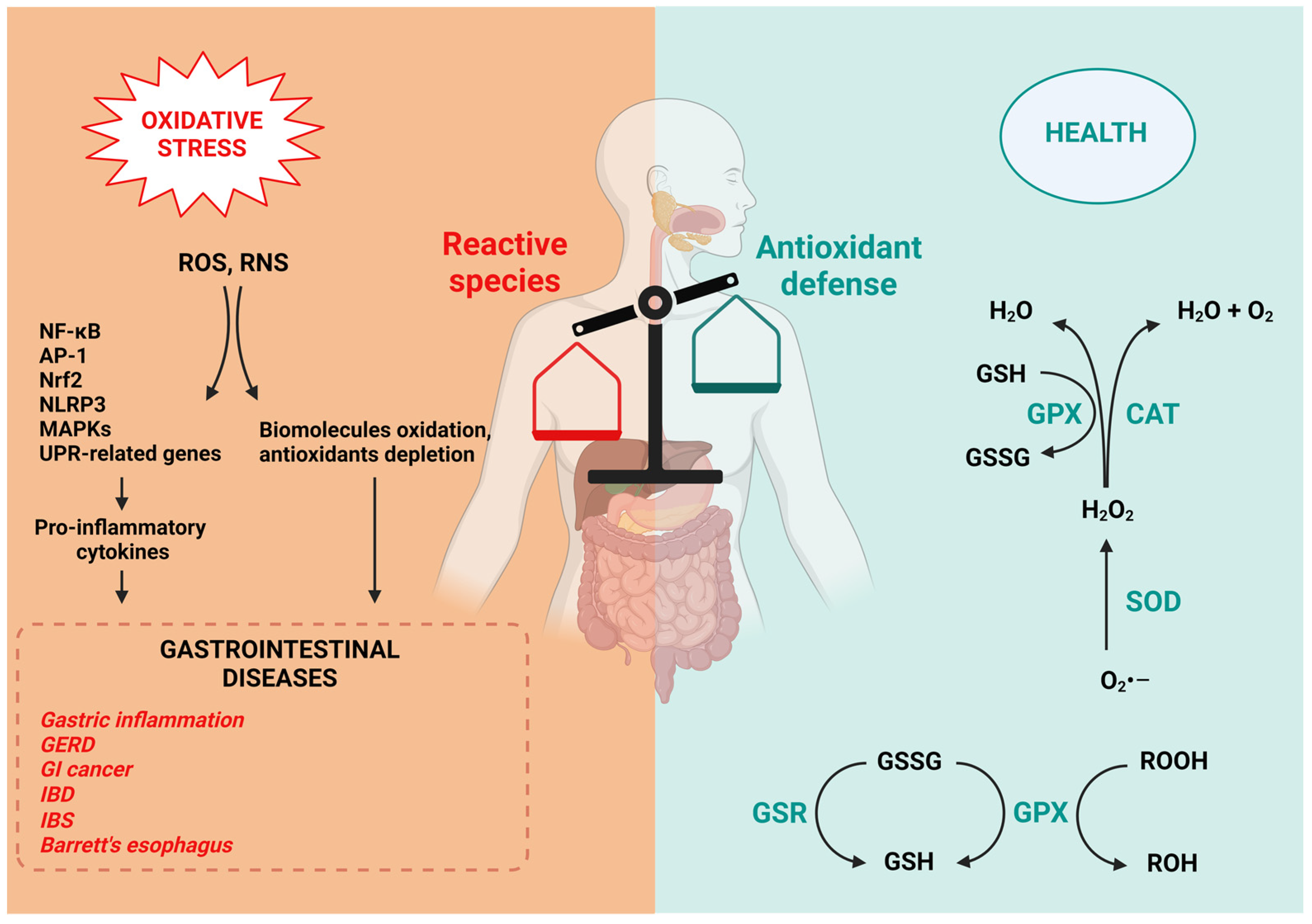
Figure 2. Unbalance between reactive species and antioxidant defense and its implications on the gastrointestinal diseases. The overproduction of free radicals due to the influence of external factors causes oxidative damage and induction of several transcription factors, which activate pro-inflammatory cytokines that together with direct oxidative damage and consequent antioxidants depletion determine the onset of GI disorders. On the other side, SOD, GPX, GSH and CAT are the lead actors that act directly in ROS-scavenging.
Superoxide dismutases are metal ion cofactor-requiring enzymes crucial for cellular protection. They catalyze the dismutation of O2•− into O2 and H2O2. In humans, there are three forms of SOD: cytosolic copper and zinc-containing enzyme (Cu-Zn-SOD), present in the mitochondrial inter-membranous space; manganese-requiring mitochondrial enzyme (Mn-SOD), present in the mitochondrial matrix; and extracellular Cu-Zn containing SOD (EC-SOD) [58]. Reduced SOD activity in the gut has been linked to gastric ulcers, while increased SOD activity has been associated with ulcer healing in patients. On the other hand, the overall activities of SODs are higher in IBD pathogenesis. This increased SOD activity serves to protect intestinal tissues from oxidative damage caused by inflammation and OS [59]. Moreover, higher levels of Mn-SOD expression have been linked to colorectal cancer. It has also been observed to be increased in normal mucosa of gastric adenocarcinoma as well as in squamous cell esophageal carcinoma tissues. In contrast, Cu-Zn-SOD levels are slightly lower in cancer tissues compared to normal tissues [60].
Glutathione peroxidase catalyzes the conversion of reduced glutathione (GSH) to oxidized glutathione (GSSG) and can reduce H2O2 to H2O and lipid hydroperoxides (ROOH) to stable alcohols. GPX, in conjunction with glutathione reductase (GSSG-R), helps maintain reduced levels of GSH, thus protecting cells from peroxide damage. There are various subtypes of GPX: GPX1, found in the cytosol of most cells; GPX2, identified in the cytosol and primarily found in the gastrointestinal tract; GPX3, present in the plasma as a glycoprotein; GPX4, located in mitochondria, where it interacts with cholesterol and lipoproteins that have been harmed by free radicals [61]. It is known that GPX isoforms are transcriptionally upregulated by oxidative mechanisms and as a result of the OS response. GPX2 expression is detected in various parts of the GI tract and is induced in gastric cancer cells. It has been observed that patients with active or remission-phase UC exhibit a significant increase in GPX activity associated with inflamed mucosa [62].
Catalase, primarily located in peroxisomes, is responsible for catalyzing the reduction of H2O2 to H2O and O2; it is predominantly distributed in the liver, kidneys, and erythrocytes [63]. CAT can be classified into the following types: typical or monofunctional catalases (e.g., mammalian catalase), bifunctional catalase peroxidases, and pseudocatalase. Mammal-type catalases function as tetramers and demonstrate comparatively lower rates of peroxidase activity. Catalase-peroxidases are characterized by their bifunctionality, serving as both catalase and peroxidase enzymes. Unlike monofunctional catalases, pseudo-catalases, also known as non-heme manganese-containing catalases, function as dimers or tetramers. These non-heme catalases comprise three well-defined and sequenced enzymes originating from various bacterial species. It is plausible that a group of pathogens linked to gastrointestinal conditions might contribute to changes in CAT expression. Diminished CAT activity has been noticed in patients with CD, as well as in patients with colorectal cancer, gastric adenocarcinoma, and H. pylori-positive gastric ulcers [64][65].
Among the endogenous non-enzymatic antioxidants, GSH is one of the most important (Figure 2). It is present at the intracellular level and is a water-soluble tripeptide that contains a thiol group derived from cysteine. GSH acts as the major soluble antioxidant in the cytoplasm, nucleus, and mitochondria. Its homeostasis is maintained through de novo synthesis, regeneration by GSSG and uptake via transport systems. GSH works in combination with GPX, GSr and GST to establish an antioxidant defense in the intestinal mucosa. Reduced GSH levels are associated with colitis, and antioxidant therapies can help restore health [7][62].
1.4. Oxidative Stress in GI Diseases
The loss of redox homeostasis is implicated in the pathogenesis of various gastrointestinal disorders (Figure 2).
Barrett’s esophagus is a metaplasia of esophageal squamous epithelium characterized by increased production of superoxide anions and subsequent lipid peroxidation, which corresponds to the inactivation of SOD. The elevation of superoxide in Barrett’s esophagus was triggered by one of the NOX isoforms, specifically NOX5. The overexpression of NOX5 is mediated by the calcium-dependent activation of Rho kinase (ROCK2) [66]. Additionally, a variant of NOX5 known as NOX5-S, which lacks calcium-binding domains, has also been observed to stimulate the generation of H2O2, resulting in DNA damage, thereby contributing to the progression from Barrett’s esophagus to adenocarcinoma [67].
As previously mentioned, NOX1 and DUOX2 play a significant role in H. pylori-induced gastric inflammation and gastric carcinoma oncogenesis. Specifically, H. pylori infection can lead to the development of peptic ulcers, which are characterized by excessive production of H2O2 and O2•− primarily derived from infiltrating leukocytes and neutrophils, as well as NOX activity. This excessive production of ROS leads to mitochondrial dysfunction, DNA alterations, and widespread tissue damage [68][69].
The advancement of IBD is affected by an intricate equilibrium of redox-sensitive pro-inflammatory pathways, like the NLRP3 inflammasome and NF-κB, and the adaptive upregulation of antioxidant enzymes, such as Mn-SOD and glutathione peroxidase 2 [70].
The excessive production of reactive oxygen species due to mitochondrial dysfunction plays a significant part in the development of IBD, including Crohn’s disease and ulcerative colitis. These conditions are distinguished by persistent inflammation of the gastrointestinal tract.
In ulcerative colitis, inflammation typically affects the mucosa and submucosa of the colon. It usually begins in the rectum and then spreads to adjacent areas, often involving the peri-appendiceal region. Pro-inflammatory cytokines, along with ROS, NOS, and increased levels of MPO, are implicated in the pathogenesis and development of ulcerative colitis. Furthermore, in ulcerative colitis, there is a loss of mucosal antioxidant defense, which contributes to inflammation and disease progression [71][72]. NO produced by iNOS stimulates the production of TNF-α in the colon. This TNF-α production leads to neutrophil infiltration by promoting the production of intercellular adhesion molecule (ICAM) and P-selectin production. The resulting neutrophil infiltration and activation contribute to tissue damage in the colon. Although both forms of IBD share similar characteristics, H2O2 and HOCl have been shown to play an important role in the pathophysiology of ulcerative colitis, while •OH and O2•− are primarily implicated in Crohn’s disease [73][74].
All areas of the gastrointestinal tract can be affected by Crohn’s disease, but the terminal ileum or perianal region is commonly involved. The features of Crohn’s disease include elevated levels of memory T cells and increased expression of major histocompatibility complex class II molecules (MHC). In the inflamed mucosa of Crohn’s disease patients, there is increased activity of XO, Mn-SOD, iNOS, and TNF-α, resulting in decreased antioxidant levels [74][75].
Furthermore, accumulated ROS could also act as secondary chemical messengers that activate intracellular signal pathways, such as p38 MAPK and NF-κB. These pathways have the ability to influence cellular processes such as cell proliferation, differentiation, and apoptosis [40].
NF-κB is a crucial transcription factor that regulates the expression of genes involved in inflammation, immune responses, and cell survival. ROS can activate the NF-κB pathway by various mechanisms, including direct oxidation and inactivation of inhibitors of NF-κB (IκBs). This leads to the release and translocation of NF-κB into the nucleus. Once in the nucleus, NF-κB promotes the expression of various proinflammatory cytokines in the intestinal epithelial cells, such as TNF-α, IL-1, IL-8, and facilitates inflammation and carcinogenesis [76].
MAPKs are highly conserved serine/threonine protein kinases, including extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38. They are involved in the regulation of cellular responses to stress and inflammation. ROS can activate MAPKs by directly oxidizing and activating upstream kinases or by modulating phosphatases that control MAPK activation. Activated MAPKs translocate to the nucleus and phosphorylate various transcription factors, thereby leading to the production of pro-inflammatory mediators and amplifying the inflammatory response in the gastrointestinal tract [77].
Recently, it has been suggested that these particulate stimuli can trigger a calcium influx mediated via transient receptor potential melastatin 2 (TRPM2), leading to activation of the NLRP3 inflammasome in irritable bowel syndrome. These findings indicate that the generation of ROS, which is a critical factor in mucosal immunity, may serve as an essential upstream event for NLRP3 inflammasome activation [78]. Activation of NF-κB can rapidly enhance the expressions of pro-IL-1β and pro-IL-18, which are then cleaved into the mature IL-1β and IL-18 by caspase-1 in the inflammasome. Subsequently, NLRP3 inflammasome initiates pyroptosis by the cleaving of gasdermin D (Gsdmd), which forms pores in the plasma membrane and acts as the executioner molecule for pyroptosis [79][80].
There is also a relationship between the unfolded protein response (UPR) and inflammation in the GI tract. The UPR is a consequence of altered endoplasmic reticulum (ER) homeostasis, which initially results in a condition called ER stress. This condition is characterized by an accumulation of damaged and improperly folded proteins within the ER lumen caused by various factors, including elevated levels of reactive oxygen species. Once the cell is affected by an ER stress, the UPR signaling pathway is activated. This pathway exerts a protective action to restore cell balance, when the stress is mild and of short duration. However, if the stress becomes too intense and prolonged, the UPR can initiate apoptotic processes that lead to cell death [81].
One of the UPR transducers, activated IRE1α, not only serves as a bifunctional enzyme but also interacts with TRAF2 (TNF receptor-associated factor 2) to activate JNK (c-Jun NH2-terminal kinase) and NF-κB. Additionally, it induces TNF-α and IL-6, which in turn further activate NF-κB, thus amplifying inflammatory responses in bowel diseases [82][83].
It is interesting to note that the degradation UPR-mediated of miR-17, a microRNA that represses the expression of the thioredoxin-interacting protein (TXNIP) by IRE1α, results in the stabilization of TXNIP. The elevated levels of TXNIP protein activate ROS overproduction and NLRP3 inflammasome. This activation leads to the cleavage of caspase-1 and the secretion of interleukin 1β, further contributing to the inflammatory response [84]. TXNIP is indeed implicated in various biological processes, including energy metabolism, insulin secretion, oxidative stress, and inflammatory response. Recent studies have highlighted the emerging role of TXNIP in the pathogenesis of complex diseases, such as metabolic disorders, neurological disorders, and inflammatory diseases [85]. Moreover, Zhenzhen et al. have demonstrated the significant impact of TXNIP on ischemia/reperfusion (I/R) injury in multiple organs, including intestinal I/R injury [86].
2. Ion Channels and Oxidative Stress
Ion channels (ICs) are integral membrane proteins that play a crucial role in regulating the ions’ flow across cell membranes. They are essential for maintaining cellular homeostasis and are involved in various physiological processes [87]. However, the function of ICs in OS conditions can be two-faced, potentially providing both benefits to the cell and contributing to pathological states.
It is remarkable to recognize that oxidative stress not only causes damage to DNA, proteins, and lipids, but it also directly regulates key pathways involved in cellular signaling controlled by ICs and protein kinases. The effect of ROS on ICs forms part of a regulatory circuit that allows cells to respond to the oxidative potential by compensating for the stress, similar to how transcription factors are activated in response to oxidative stress, leading to the expression of antioxidant components. Additionally, activation by OS can serve as a second messenger in signaling cascades driven by changes in ion channel activity in response to hormones and neurotransmitters. However, excessive production of reactive species and constant activation of OS lead to alterations of ICs that extend beyond ion fluxes and can impact diverse cellular processes, including membrane excitability, signal transduction, neurotransmission, muscle contraction, hormone secretion, cell proliferation, and apoptosis. Thus, OS-induced dysfunction of ICs and transporters can contribute to the pathogenesis of various disorders, including cardiovascular diseases, neurodegenerative conditions, cancer, and metabolic and liver diseases [88][89].
Therefore, understanding the interplay between ion channels and oxidative stress is essential for unraveling the mechanisms underlying cellular responses to oxidative stress and their implications in disease pathophysiology.
The presence of ROS/RNS can directly interact with ion channels and induce post-translational modifications, such as oxidation, nitrosylation, or nitration of specific amino acid residues. These modifications occur on amino acids that are particularly susceptible to modification by ROS/RNS due to the presence of sulfur atoms, hydroxyl groups, or aromatic rings [90].
Several types of ICs are known to be affected by oxidative stress. For instance, voltage-gated ion channels, such as sodium, potassium, and calcium channels, can experience altered gating properties and ion selectivity. Oxidative stress can also modulate ligand-gated ion channels, including receptors for neurotransmitters and hormones, thereby affecting their sensitivity and signaling pathways [88]. Additionally, OS can impact intracellular calcium homeostasis by altering the activity of calcium release channels in the ER (e.g., ryanodine receptors) and calcium uptake channels in the plasma membrane (e.g., store-operated calcium channels) [91].
Ion transporters, including those responsible for sodium, potassium, calcium, and chloride ion transporters, are also influenced by oxidative stress. ROS can affect the activity and expression levels of these ICs, leading to disruptions in ion gradients and cellular ionic balance. Moreover, OS can impair the function of sodium-potassium ATPase, compromising ion homeostasis and cellular energy metabolism [92].
2.1. Ion Channels Involved in Oxidative Stress-Related GI Diseases
Ion channels play a critical role in the physiopathology of the gastrointestinal tract. Intestinal epithelial cells use ion channels present on the submucosal nerves and enterochromaffin cells to transport ions which in turn drive fluid secretions. ICs also regulate the normal function of the GI smooth muscle, which is critical for optimal motility. In this regard, in tissues that require coordinated contractions (such as smooth, cardiac, and striated muscle), electrical depolarizations play a fundamental role in ensuring proper electromechanical coupling. Therefore, the coordination of electrical activity is so well tuned that even small perturbations can lead to functional abnormalities [93][94].
The chronic inflammation present in some gastrointestinal diseases, such as IBD, may contribute to increased oxidative stress in the gut. OS can lead to dysregulation of ICs in the gastrointestinal tract, by altering electrolyte transport, as well as increasing the secretion of mucus and water, and reducing absorption of nutrients, which contribute to the development of inflammation and tissue damage [95]. There are several types of ion channels involved in gastrointestinal pathologies, including chloride, calcium, potassium, and sodium channels. The main ones are disclosed below.
2.2. Calcium Channels
Calcium channels are critical for smooth muscle contraction, gene expression, neurotransmitter release, and hormone secretion in the gastrointestinal tract [96].
2.2.1. Voltage-Gated Ca2+ Channels
The family of voltage-gated Ca2+ channel (CaV) consists of five main subgroups: L-type (CaV 1.1–1.4), N-type (CaV 2.2), P/Q-type (CaV 2.1), R-type (CaV 2.3), and T-type (CaV 3.1–3.3) Ca2+ channels [97].
CaV channels were among the first Ca2+ channels to be discovered as being responsive to changes in cellular redox status. Voltage-sensitive calcium channels L-type have been described in smooth muscle cells (SMCs) and interstitial cells of Cajal (ICCs), known to be the pacemaker cells and control the contraction of the smooth muscle cells of the gastrointestinal tract [98][99].
Calcium influx in gastrointestinal smooth muscle is primarily mediated by the voltage-gated L-type Ca2+ channel, which is responsible for the upstroke of the action potential. The Ca2+ channel is a multimeric complex consisting of a central pore-forming and voltage-sensing α subunit along with auxiliary β and α2δ subunits [100]. Links have been reported between L-type calcium channel activity, oxidative stress, and gastrointestinal pathologies, including IBD [101].
In the inflamed colon, the circular smooth muscle cells exhibit a decrease in the expression of L-type calcium channels on their membranes. This down-regulation potentially leads to a reduction in calcium influx. Despite this decrease, the functional and pharmacological properties of the channels appear to be normal. However, acetylcholine, a neurotransmitter involved in muscle contraction, fails to activate these channels in the inflamed cells. This inability to activate the channels may be due to additional defects in the receptor signaling cascade upstream. The reduced expression of L-type calcium channels could dampen the contractions of circular smooth muscle in the inflamed colon, contributing to the motility and digestion abnormalities observed during inflammatory disorders [102][103][104][105].
Furthermore, in the context of OS, the expression of tyrosine-nitrated calcium channels on the plasma membrane is increased, which hinders the binding of Src tyrosine kinase. The enhanced production of nitric oxide leads to the formation of peroxynitrite, which oxidizes tyrosine residues and results in the formation of 3-nitrotyrosine. This OS-induced modification alters the gating kinetics of calcium channels, leading to decreased calcium influx [106][107].
The regulation of calcium channels is crucial for the process of calcium-induced gene transcription and one of the transcription factors closely associated with the activation of L-type calcium channels is the cyclic AMP response element-binding protein (CREB). Following cell depolarization, CREB undergoes specific phosphorylation at the Ser133 site within the nucleus. Oxidative stress impacts excitation-transcription coupling, resulting in reduced depolarization-mediated expression of phospho-CREB due to tyrosine nitration of the calcium channel. The particular post-translational modification is noteworthy, as S-nitrosylation enhances L-type calcium currents, whereas tyrosine nitration predominantly occurs in colon inflammation [108][109][110].
2.2.2. Calcium-Permeable Non-Selective Ion Channels
The transient receptor potential (TRP) channels comprise a superfamily of non-selective calcium-permeable ion channels. In particular, they are a class of cationic channels that can act as signal transducers by modulating membrane potential or intracellular calcium concentration. This ICs family can be divided into seven subgroups: TRPC (Canonical), TRPV (Vanilloid), TRPM (Melastatin), TRPA (Ankyrin), TRPML (Mucolipin), TRPP (Polycystic), and TRPN (NOMPC-like). All seven members of the subfamily share a similar structural organization. They consist of six transmembrane spanning domains (S1–S6), with the pore region formed by hydrophobic residues between S5 and S6. Additionally, they possess three to four ankyrin repeats, coiled-coil domains in the N- and C-terminus, a C-terminal proline-rich region, a Calmodulin/IP3 binding region and a 25-amino acid motif, known as TRP domain. This TRP domain, loosely conserved among almost all TRP channels, is located at the C-terminal end of the sixth transmembrane segment [111].
These channels are involved in multiple cellular functions, including cell death, chemokines production, pain sensation and thermosensation. Their activation mechanisms involve not only the direct detection of temperature and/or chemical compounds and changes in extracellular or intracellular ion concentrations but also the detection of second messengers. Some TRP channels, including TRPM2, TRPM7, TRPC5, TRPV1, and TRPA, sense reactive species either indirectly through second messengers or directly via oxidative modification of cysteine residues [112][113].
TRP channels in the GI tract function as molecular detectors of chemical and physical signals and act as secondary responders to G protein-coupled receptors (GPCRs) or ion channel receptors, thereby regulating calcium balance. These channels are expressed not only in the sensory neurons of the ganglia and enteric nervous system that innervate the GI tract but also in non-neuronal cells such as intestinal epithelial cells, enteroendocrine cells, and immune cells. Among the TRP channel subfamilies, TRPV, TRPA, and TRPM have been identified as particularly relevant in inflammatory bowel disease. They can be activated by various endogenous stimuli and have been extensively studied as potential targets for therapeutic drugs. Alterations in the expression of these TRP channels have been observed in patients with IBD and animal models of colitis, indicating their involvement in the disease [114][115][116].
TRPM2
The TRPM2 ion channel exhibits permeability to divalent ions such as Ca2+ and Mg2+, as well as monovalent ions including Na+ and K+. It contains an enzymatic region known as the NudT9-H domain, which possesses ADP-ribose (ADPR)-hydrolase activity. TRPM2 is expressed in various gastrointestinal cells, including enterocytes of the intestinal epithelium, enteroendocrine cells, pancreatic β-cells, smooth muscle cells, and some immune cells present in the intestinal mucosa. Under normal physiological conditions, TRPM2 channels play a crucial role in immune modulation by regulating the release of various cytokines, such as TNF-α, IL-6, IL-8, and IL-10, in human monocytes. They are also involved in the maturation and chemotaxis of dendritic cells, which are essential for immune responses. In addition to their role in immune modulation, TRPM2 channels are also implicated in other functions such as regulating endothelial permeability and insulin secretion [117][118].
The expression of TRPM2 in various gastrointestinal cells suggests its involvement in the control of gastrointestinal function and responses to pathological conditions.
One notable characteristic of TRPM2 is its modulation by oxidative stress. It is hypothesized that oxidative/nitrosative stress triggers a biochemical pathway within mitochondria, leading to the production of ADP-ribose. This molecule acts as a diffusible second messenger that can activate TRPM2 channels located on the cell’s plasma membrane [119][120].
Under OS, H2O2 promotes the generation of intracellular ADPR in mitochondria. This event leads to the activation of two enzymes in the nucleus: PARP-1 (poly ADP-ribose polymerase) and poly ADP-ribose glycohydrolase (PARG). PARP-1 binds to damaged DNA and catalyzes the cleavage of NAD+ into nicotinamide and intracellular ADPR. Both H2O2 and ADPR can synergistically activate TRPM2. Intracellular ADPR binds to the C-terminal NudT9-H domain of the channel, leading to increased calcium influx. This influx triggers the phosphorylation of Pyk2, which enhances Ras-dependent ERK activation. Amplified ERK activates the transcription of the CXCL8 gene, inducing the nuclear translocation of RelA, an NF-κB subunit in human monocytes. This cascade ultimately leads to the upregulation of pro-inflammatory cytokines [121][122].
Ca2+ influx through TRPM2 also activates both extrinsic and intrinsic cell death pathways, resulting in the activation of caspase-3 and the cleavage of PARP [123].
Structural properties play a significant role in the oxidative modulation of TRPM2. A shorter isoform of TRPM2 (TRPM2-S) lacking specific transmembrane domains has been identified. TRPM2-S directly interacts with the full-length isoform (TRPM-L) and acts as a dominant negative mutant, inhibiting TRPM2-mediated Ca2+ influx. Interestingly, co-transfecting TRPM2-L and TRPM2-S suppresses H2O2-induced cell death by inhibiting OS-induced TRPM2 activity. These findings suggest that the dominant negative effects of TRPM2-S protect cells from oxidative damage [124][125].
In the gastrointestinal tract, TRPM2 channels are expressed in mucosal macrophages, mast cells and enteric neurons and they have been shown to contribute to the progression of experimental colitis and food allergy in mice. Notably, TRPM2 deficiency suppresses the exacerbation of inflammation in a mouse model of dextran sulfate sodium (DSS)-induced colitis. TRPM2 has been suggested to play an important role in the colon, leading to the production of cytokines and chemokines (such as CXCL2) by macrophages, neutrophils, and T lymphocytes [126][127].
However, the physiological and pathological roles of TRPM2 in the gastrointestinal tract are not well understood. It is hypothesized that TRPM2 plays an important role in visceral nociception and the development of hypersensitivity because several lines of evidence indicate that TRPM2 channels are likely expressed in neurons [128][129].
Matsumoto et al. have founded that intestinal manipulation activates resident muscle macrophages via TRPM2, leading to the release of inflammatory cytokines and chemokines, which in turn promote the infiltration of macrophages and neutrophils into the muscle layer, resulting in dysmotility. Therefore, TRPM2 could be an interesting target for the treatment of dysmotility associated with postoperative ileus [130].
TRPM2 is also found in pancreatic β-cells, where its activity has been linked to insulin secretion and H2O2-mediated apoptosis of insulin-secreting cells, suggesting a role of TRPM2 in diabetes [131].
TRPV1
TRPV1 is primarily found in sensory nerve fibers throughout the gastrointestinal tract, including the esophagus, stomach, small intestine, and colon. It is also expressed in certain non-neuronal cells such as epithelial cells, smooth muscle cells, and immune cells within the gastrointestinal tract [132][133]. TRPV1 is a channel that responds to heat, capsaicin and acidic pH, and it can be upregulated or sensitized by pro-algesic pathways associated with functional GI disorders. In patients with conditions such as IBS and non-erosive reflux disease, upregulation of TRPV1, even in the absence of overt inflammation, has been observed [134][135].
During OS, when TRPV1 is exposed to hydrogen peroxide, it exhibits enhanced responses to capsaicin, acid, and receptor phosphorylation, indicating sensitization. This sensitization occurs through the formation of inter-cysteine disulfide bonds within the cytoplasmic termini of TRPV1. Several cysteine residues, including C772 and C783, play a critical role in this sensitization process. Oxidized TRPV1 receptors are more resistant to desensitization induced by capsaicin, while desensitized receptors can be reactivated by strong OS. This oxidative modulation of TRPV1 operates independently of phosphorylation, desensitization, and extracellular pH, and it amplifies the channel’s response to noxious stimuli [135][136].
Furthermore, TRPV1 is also expressed on the plasma membrane of esophageal epithelial cells, and its activation by capsaicin and acid can induce significant production of IL-8 suggesting that TRPV1 activation on esophageal epithelial cells may contribute to esophageal inflammation through cytokine release. In a study, it was observed that the activation of TRPV1 receptors by capsaicin led to a significant increase in the production of ROS within Het1A cells. Interestingly, when a TRPV1 antagonist was applied, the elevated levels of ROS-modified proteins induced by capsaicin treatment were blocked. These findings suggest that capsaicin may specifically trigger ROS production through a TRPV1-dependent pathway [137][138][139].
TRPV1 activation in esophageal epithelial cells can specifically induce ROS production, which can potentially modify intracellular biomolecules, including TRPV1 itself. Bile acids and low pH can induce oxidative stress and DNA damage in esophageal tissues and cells by, suggesting the possibility that acid-induced OS through TRPV1 activation may occur in esophageal epithelial cells [140].
TRPV1 is involved in gastroesophageal reflux disease, a gastrointestinal disorder associated with the malfunctioning of the lower esophageal sphincter (LES), leading to the reflux of stomach acid and digestive juices into the esophagus. While the primary cause of GERD is the weakening of the LES, the involvement of TRPV1 receptors has also been observed [138][141].
TRPV1 receptors, when activated, can contribute to the sensation of heartburn and chest discomfort experienced by GERD patients. Acidic substances, such as stomach acid, can stimulate and activate TRPV1 receptors in the esophagus, leading to the perception of pain and irritation. The activation of TRPV1 receptors in the esophagus may also enhance sensitivity to pain and contribute to the development of esophageal hypersensitivity, which is a characteristic feature of GERD.
Research suggests that TRPV1 antagonists, which inhibit the activity of TRPV1 receptors, could potentially provide therapeutic benefits in managing GERD symptoms. By blocking TRPV1 receptors, these antagonists may help decrease esophageal sensitivity to acid reflux and alleviate the discomfort and pain associated with it [142][143].
TRPA1
The ion channel TRPA1 is recognized for its excitatory characteristics and is thought to be involved in diverse sensory processes, including cold perception, burning sensation and discomfort associated with acid reflux, as well as inflammation pain in the context of IBD. While it is well-established that TRPA1 is expressed in a subset of sensory neurons, particularly nociceptors, its functional role in the gastrointestinal tract, especially in non-neuronal cells, such as enteroendocrine cells, remains largely unexplored. TRPA1 exhibits significant expression in both human and rat enteroendocrine cells, suggesting that it may function as a sensor molecule in these cells to regulate gastrointestinal functions [144][145][146].
TRPA1 is found mainly on the cell plasma membrane and presents in the intracellular portion of cysteine residues, which have been identified as key players in its activation. Agents that modify cysteine, such as N-methyl maleimide, have been shown to activate TRPA1, while agents that reduce cysteine have the opposite effects [147][148]. These findings suggest that cysteine oxidation in TRPA1 may be important for its activation. Considering that H2O2 induces oxidative modification of protein cysteine residues, it is possible that H2O2 may activate TRPA1 inducing the activation of sensory neurons in the pain pathway [149]. This mechanism may be related to the neurogenic inflammation in the gastrointestinal tract in gastroesophageal reflux disease. Experimental evidence revealed that berberine attenuated neurogenic inflammation in the gastrointestinal tract mainly by suppressing the production of substance P (SP) [150]. Additionally, Zou et al. showed that berberine can also suppress the upregulation of TRPA1 induced by airway hyperresponsiveness (AHR), a common symptom observed in GERD patients [151].
Nrf2 is a transcription factor that participates in the regulation of cellular response to oxidative stress. It has been widely reported that Nrf2 is dissociated from the Cul3-kelch-associated ECH-associated protein 1 (Keap1)-Nrf2 complex by ROS, after which Nrf2 enters the nucleus to mediate the expression of TRPA1. Given that Nrf2 can mediate the expression of TRPA1, and that ROS are activators of TRPA1, it has been hypothesized that ROS not only activate TRPA1 but also regulate TRPA1 expression via Nrf2. Furthermore, the influx of Ca2+ through TRPA1 activates the ERK and AKT signaling pathways leading to apoptosis by activating calcium/calmodulin-dependent kinase II (CaMKII), a serine/threonine protein kinase. CaMKII undergoes autophosphorylation in the presence of Ca2+ and CaM, and it subsequently activates various downstream effectors. Hydroxyl radical scavengers have shown effectiveness in alleviating ROS or oxidative stress-related diseases by blocking TRPA1 activation. Additionally, TRPA1 has been found to be upregulated in various cancer cells, including colorectal cancer. This suggests a potential role of TRPA1 in cancer development and progression [152][153][154][155][156].
Moreover, elevated cytosolic Ca2+ concentrations induce Ca2+ uptake into the mitochondrial matrix in cells, leading to depolarization of the inner mitochondrial membrane and alteration of the outer membrane permeability. These events trigger the release of cytochrome c and Apaf-1-dependent activation of the apoptosome, which is a multi-subunit protein complex that serves as a platform for caspase activation, ultimately leading to apoptosis. An increase in intracellular Ca2+ levels also leads to membrane depolarization and subsequent activation of UPR [156][157][158].
2.3. Chloride Channels
Chloride ion channels are important regulators of fluid and electrolyte transport in the gastrointestinal tract, and their dysfunction has been implicated in the pathogenesis of several GI disorders. There are several types of chloride channels expressed in the gastrointestinal tract, including the cystic fibrosis transmembrane conductance regulator (CFTR), calcium-activated chloride channels (CaCCs) and Chloride intracellular channel 1 (CLIC1).
2.3.1. Cystic Fibrosis Transmembrane Conductance Regulator
Cystic fibrosis is a genetic disorder characterized by the abnormal folding of the CFTR protein. One of the most common mutations associated with cystic fibrosis is the deletion of phenylalanine at position 508 (ΔF508) in the CFTR protein. These mutations impair the function of CFTR, leading to disrupted chloride and fluid transport across epithelial cells in various organs, including the lungs, pancreas, and gastrointestinal tract. In the latter, CFTR dysfunction can result in reduced chloride secretion and increased sodium and water absorption. This imbalance leads to the production of thick and sticky mucus that obstructs the pancreatic ducts, bile ducts, and small intestine, causing complications and impairing normal digestive processes [159][160].
In CF, misfolded CFTR proteins are retained within the ER and undergo rapid degradation by the ERAD (ER-associated protein degradation). This holds true for the most common mutation, ΔF508. The retention of misfolded proteins can lead to ER stress, which activates UPR with the purpose of regulating the protein load within the ER. This involves increasing the expression of chaperones and limiting overall protein synthesis in cells. Moreover, UPR limits the endogenous expression of CFTR (wild type and ΔF508) by the activation of its transducer ATF6 (activating transcription factor 6) to limit gastrointestinal tract manifestations of cystic fibrosis, including mucous inspissation, dysmotility, constipation, and GERD [161][162][163].
Oxidative stress can modulate CFTR channel activity through direct oxidation of cysteine residues or via downstream signaling pathways. OS modulates intracellular signaling pathways mediated by protein kinase A (PKA) and protein kinase C (PKC), which are known regulators of CFTR function. ROS-mediated activation of these pathways can modulate CFTR phosphorylation and alter its activity and localization. In addition, CFTR is involved in maintaining cellular redox balance by regulating the transport of GSH. CFTR dysfunction in CF may result in reduced GSH levels and compromised antioxidant defenses, leading to increased oxidative stress in the GI tract [164][165][166].
The interplay between CFTR dysfunction and OS contributes to the pathogenesis of various GI disorders. Conditions such as Crohn’s disease and ulcerative colitis are characterized by increased OS and impaired CFTR function. The elevated oxidative stress in these conditions exacerbates inflammation, disrupts ion and fluid transport, and compromises the integrity of the intestinal barrier [167][168].
The regulation of CFTR expression in oxidative stress conditions involves the HIF-1/Nrf2 pathway. Nrf2, which is normally located in the cytoplasm, translocates to the nucleus in response to cellular stress. In the nucleus, Nrf2 activates target genes by binding to antioxidant response elements. However, the effect of Nrf2 on CFTR expression is context-dependent and varies based on tissue type, cell conditions (normoxic or hypoxic), and possible cancer stage. Nrf2 has been reported to both positively and negatively regulate CFTR expression, and its downregulation is observed in patients with CF and CF knockout mouse models. The CFTR gene contains CREs (cAMP response elements) within a topologically associating domain. Under normoxic conditions, repressor proteins, including BACH1, bind to the −44 kb ARE located in the TAD, leading to CFTR repression. However, oxidative stress, induced by compounds like sulforaphane (SFN), displaces repressors and activates CFTR expression in airway epithelial cells [169][170][171][172].
The regulation of CFTR expression in intestinal epithelial cells, especially colorectal cancer (CRC) cells, remains less understood. Stress conditions like gut ischemia, hypoxia, dysbiosis, and inflammation have been linked to CFTR dysregulation. Hypoxia and HIF-1 signaling repress CFTR expression in CRC cells, which is commonly observed in epithelial cancers [173][174][175].
The role of CFTR in carcinogenesis is complex and may vary depending on the tissue and cancer context. Loss of CFTR may protect CRC cells from ROS-induced apoptosis by retaining antioxidants such as GSH, thereby promoting cell survival. Initial evidence suggests that loss of CFTR in CRC cells enhances their survival when exposed to oxidative stress. Overall, the regulation of CFTR expression in OS conditions involves intricate molecular pathways, including the HIF-1/Nrf2 pathway. The relationship between CFTR, oxidative stress, and carcinogenesis is multifaceted and context-dependent, requiring further investigation to fully understand its implications in different tissues and cancer types [176][177][178].
2.3.2. Calcium-Activated Chloride Channels
Calcium-activated chloride channels are extensively expressed in various tissues and implicated in physiological processes such as sensory transduction, epithelial secretion, and smooth muscle contraction. CaCCs are mainly found on the plasma membrane of the epithelial cells of the gastrointestinal tract and of the interstitial cells of Cajal. Activation of CaCCs enables the efflux of chloride ions from cells. This process is crucial for facilitating digestion and maintaining appropriate hydration levels within the GI tract [179].
CaCCs are heterogeneous groups of ligand-gated chloride ion channels that include proteins from several structurally diverse families: accessory chloride channel (CLCA), bestrophin (BEST), and calcium-dependent chloride channel anoctamin (ANO or TMEM16) channels. The TMEM16 is highly expressed in the human gastrointestinal interstitial cells of Cajal. Some CLCA isoforms, both human and mouse, may have roles as tumor suppressors, particularly in mammary carcinoma. One member of the CLCA family, mCLCA6, is highly expressed in the intestine and stomach, with trace amounts in the spleen and liver, while mCLCA3 overexpression has been linked to mucus overexpression in cystic fibrosis. Overall, CaCCs are activated by an increase in intracellular calcium levels, and their function is modulated by OS [180][181][182].
Oxidative stress can directly influence CaCCs through several mechanisms in the GI tract: ROS induces oxidative modifications of CaCC proteins, such as the oxidation of critical cysteine residues. These modifications can alter the channel’s gating properties, stability, and trafficking, leading to aberrant chloride transport and fluid secretion. CaCCs are activated by intracellular calcium concentration and phosphorylation of Ca2+/calmodulin protein kinase. The intracellular hydroxyl radicals directly increase the activity of CaCC channels, going to targeting, reversibly or irreversibly, exposed cysteine -SH residues [183][184].
Dysregulation of CaCCs due to oxidative stress has been implicated in various GI disorders. For instance, in inflammatory bowel disease, increased OS can lead to CaCC dysfunction, contributing to altered chloride transport and excessive fluid secretion. This can result in diarrhea and electrolyte imbalances [185].
2.3.3. Chloride Intracellular Channels
CLIC1 is a member of the CLIC protein family and plays an important role in the transport of chloride ions across cellular membranes. Emerging evidence suggests that CLIC1 is involved in various cellular processes, including cell proliferation, apoptosis, and ion homeostasis [186][187].
There is still controversy regarding the functional expression of CLIC1. For most researchers actively working on protein properties, CLIC1 functions as a selective chloride channel with unique characteristics that are transiently expressed on the membrane in response to specific stimuli. However, since it is considered unlikely for a hydrophilic protein to undergo drastic changes enabling insertion into the membrane and formation of an ion channel, not everyone considers it as such. Furthermore, the recordings made on the membrane could potentially be attributed to modulation by CLIC1 on other resident chloride channels in the membrane. In this latter case, CLIC1 would function as a second messenger or an additional functional subunit [188][189].
Over the past 10 years, several research groups have demonstrated, using different experimental approaches, that CLIC1 could realistically form an ion pathway. According to this view, CLIC1 would be an example of an ion-permeable protein transiently expressed in the cell membrane. However, the mechanism of protein anchoring and insertion into the lipid bilayer is still unknown [190].
CLIC1 has been shown to be involved in cell cycle regulation being detected on the plasma membranes of cells in the G2/M phase. During this period, the current density is approximately twice that recorded in the G1/S phase, and inhibition of CLIC1 function prolongs the average cell cycle time in cell culture. It is known that there are oxidative fluctuations during the cell cycle that drive cells through all phases of the cell cycle. Therefore, it is not surprising that CLIC1 is hyperactivated in all diseases involving OS, including GI tumors. The behavior of CLIC1, migrating to the plasma membrane in response to changes in the redox state of cells, given the protein’s high affinity for GSH, suggests that CLIC1 should be explored as a potential therapeutic target [191][192].
CLIC1 is overexpressed due to increased ROS, particularly hydrogen peroxide. The overexpressed channel suppresses the nuclear translocation of Nrf2 and cellular antioxidant capacity, promoting cellular senescence and endothelial dysfunction. The main characteristics of Nrf2 are to some extent mimicked by Nrf2-dependent genes and their proteins, including heme oxygenase-1 (HO-1). HO-1 exerts beneficial effects through protection against oxidative damage, regulation of apoptosis, modulation of inflammation, and contribution to angiogenesis. Conversely, Nrf2/HO-1 activation is essential in the mechanism by which CLIC1 activation is blocked and in the expression of antioxidant systems. In fact, the nuclear translocation of Nrf2 is blocked by CLIC1 overexpression but enhanced by the treatment with an IAA94 inhibitor or CLIC knockdown [193][194][195].
Upregulation of the CLIC1 protein has been linked to the progression of gallbladder and gastric cancers, where it promotes cell proliferation via regulation of MAPK/AKT pathways and facilitates fibroblast formation. In fact, Li et al. demonstrated that the expression levels of integrin α3 (ITGα3), integrin αv (ITGαv), integrin β1 (ITGβ1) and Bcl-2 decreased in gastric tumor cells after CLIC1 knockdown, as well as AKT-phosphorylation, ERK-phosphorylation, and p38-phosphorylation. Therefore, it has been hypothesized that CLIC1 may play an important role in the initiation and progression of gastric cancer and its mechanism may be related to the regulation of integrin family proteins, leading to the sequential regulation of PI3K/AKT, MAPK/ERK and MAPK/p38. Furthermore, the upregulation of CLIC1 protein is associated with the metastatic capacity of colorectal cancer cells, where it has been demonstrated to control cell volume and ROS levels [196][197].
2.4. Potassium Channels
The remarkable diversity of genes encoding K+ channels represents the largest group of ion channels in the human genome. These genes often have splice variants, undergo post-translational modifications, or form complexes with regulatory subunits, further expanding the functional variability of K+ channels. This variability underscores the significance of fine-tuning K+ conductance and raises questions about additional functions these channels may serve. Voltage-gated, Ca2+-activated, inward rectifier channels, and 2P-domain channels are the general classifications of K+ channels [198][199][200].
While sodium (Nav) channels are typically associated with the rapid depolarization of excitable cells, voltage-gated K+ (Kv) channels are responsible for the repolarization phase during an action potential. In non-excitable cells like those in the GI epithelium, these channels primarily function to hyperpolarize the PM. This negative membrane potential is essential for Ca2+ signaling, intracellular pH regulation, and cell volume control. Due to their wide-ranging impact, K+ channels are implicated in various cellular and tissue functions, including cell proliferation, differentiation, contractility, circadian rhythms, wound healing, cell cycle progression, autophagy, metabolism, angiogenesis, stem cell dynamics, making them vital for cellular functions in various tissues, such as smooth muscles, pancreatic β-cells, myocardium, and neurons. However, the mechanisms underlying the loss of control over K+ channels are still not fully understood [187][201].
Research indicates that the functional characteristics of the human ether-related gene (HERG) potassium channels may be affected by ROS, possibly through interactions involving cysteine or histidine residues. Methionine is also susceptible to oxidation; when an oxygen atom is added to its sulfur-containing region, it forms methionine sulphoxide (MetO), which possesses distinct physicochemical properties compared to methionine. Protein oxidation in methionine residues frequently leads to functional impairments, including a reduction in cellular electrical excitability [202][203].
2.4.1. The ATP-Sensitive Potassium Channels
In the maintenance of plasma membrane potential, K+ channels play a vital role. They work in conjunction with transporters such as the Na+/K+-ATPase, H+/K+-ATPase, and the Na+-K+-2Cl− cotransporter (NKCC) to facilitate the exit of K+ ions from the cell, thereby maintaining a net negative charge at the membrane potential. This hyperpolarized membrane potential is crucial for driving the active transport of various molecules against their concentration gradients. Particularly in the GI epithelium, which requires continuous transport of water, electrolytes, and nutrients, K+ channels are significant [199][200].
The ATP-sensitive potassium channels consist of regulatory sulfonylurea subunits (SUR1, SUR2A, or SUR2B) along with pore-forming potassium channel subunits (Kir6.1 or Kir6.2). They are influenced by physiological and pathophysiological factors, including hypoxia, hyperglycemia, ischemia, and OS, enabling them to regulate cellular excitability based on metabolic conditions [204].
These channels have various functions in GI tissues: regulation of insulin secretion, protection against cellular stress, and adaptation to metabolic changes [205][206].
ROS can either decrease or increase KATP channel activity, depending on the tissue and the specific isoforms expressed. Redox regulation has been linked to the N-terminus of the Kir6.2 subunit, particularly Cys42. This mutation made the channel unresponsive to sulfhydryl reactive agents, suggesting that the Kir subunit is a possible site for redox regulation. The modification of Cys42 through sulfhydryl alters the channel allosterically, rather than causing direct pore blockage. NO activates K+-ATP channels through S-nitrosylation of the SUR1 subunit, while S-glutathionylation of Cys176 in the Kir6.1 subunit inhibits the Kir6.1/SUR2B channel. Protein S-glutathionylation is promoted by oxidative or nitrosative stress. These post-translational modifications are associated with OS and can impact channel function. The effects of NO on KATP channel activity are complex. Stimulation and inhibition of channel activity have been reported depending on the concentration of NO. Indirect effects of NO on KATP channels have been ascribed to interference with mitochondrial metabolism and the cGMP/protein kinase G pathway, while high concentrations of NO can inhibit KATP channels directly [207][208][209].
Interestingly, the loss of KATP channel activity in β-cells provided partial protection against apoptosis induced by hydrogen peroxide and nitric oxide. This protection appeared to be due to the upregulation of antioxidant enzymes, including SOD, GPX and CAT, which help to detoxify ROS and RNS. This upregulation of antioxidant enzymes seemed to be a result of increased calcium levels in the cytosol and mitochondria, which occurs when KATP channels are genetically or pharmacologically inhibited [210][211].
The enhanced oxidative stress associated with type 2 diabetes mellitus contributes to disease pathogenesis. ROS and RNS can cause oxidative stress, which affects the function of β-cells in the pancreas leading to apoptotic cell death. Previous studies have demonstrated that ROS/RNS can impact the activity of ATP-sensitive potassium channels in β-cells by inhibiting the production of ATP in the mitochondria. Both hydrogen peroxide and alloxan, a diabetogenic agent that is believed to generate H2O2, can open KATP channels and change the electrical potential of the β-cells membrane. Oxidative damage increases the production of ROS in mitochondria, and these ROS can trigger the opening of a pore in the mitochondrial membrane, generating a collapse of the membrane potential and depletion of ATP [212][213].
There is a growing body of evidence indicating that hydrogen sulfide (H2S), similar to nitric oxide and carbon monoxide, functions as a signaling molecule that plays a crucial role in both normal physiology and the development of various diseases. In the case of IBD, H2S has been found to have a protective role in the gastrointestinal tract.
In experimental models of colitis, it has been observed that colonic inflammation leads to increased activity of ATP-sensitive potassium channels in the smooth muscle cells of the colon. The precise molecular mechanisms underlying this alteration are not yet fully understood, but it appears that these KATP channels may be potential targets for the effects of H2S. However, oxidative stress is implicated in this process. H2S can modify the KATP channels by S-sulfhydrating cysteine residues, particularly in the SUR2B subunit. This modification enhances the activity of KATP channels in the smooth muscle cells of the colon, thereby improving colonic function and aiding in the resolution of inflammation [214][215][216].
During colonic inflammation, there is an increased production of H2S, which contributes to the resolution of the inflammatory state. Animal models of colitis have shown that inhibiting KATP channels leads to significant mortality, underscoring the importance of these channels in the context of ulcerative colitis and highlighting their potential as therapeutic targets.
In the DSS colitis mouse model, it has been observed that the response to the KATP channel agonist lemakalim is significantly enhanced in inflamed smooth muscle cells, along with a down-regulation of calcium currents. Further investigation revealed increased bursting activity of individual KATP channels in inflamed mice. The major isoforms of KATP channels in mouse colonic smooth muscle are Kir6.1 and SUR2B. Gene expression analysis showed up-regulation of Kir6.1 mRNA and down-regulation of SUR2B mRNA during inflammation. However, the impact of these changes on protein expression remains unclear, as protein expression was not assessed [217][218].
2.4.2. Kv Channels
Kv channels are present in gastrointestinal smooth muscle cells, contributing to the regulation of muscle excitability and contraction. These channels regulate the resting potential and action potential of smooth muscle cells, affecting the motility of the gastrointestinal tract. In enteric neurons, Kv channels influence the generation and propagation of electrical impulses [219].
Some endocrine cells found in the gastrointestinal tract, such as the Langerhans cells in the intestine, express Kv channels. These channels can influence the secretion of hormones and the regulation of chemical signals in the gastrointestinal environment. Since Kv channels are implicated in the regulation of gastrointestinal motility, their alterations may contribute to motor dysfunctions related to functional abdominal pain and intestinal motility disorders.
Kv consists of four α subunits; each α subunit contains six transmembrane segments and contributes to the formation of the conductive pore for potassium ions. In addition to the α subunits, Kv receptors may also include accessory or regulatory subunits that influence their activity. Some examples of common accessory subunits are β and γ subunits, which can regulate the function and properties of voltage-gated potassium channels.
The auxiliary subunits, known as Kvb subunits, interact with the pore-forming α -subunits and influence the inactivation rate of Kv channels. Kvb1 and 3 possess a long N-terminus that induces channel inactivation, whereas Kvb2 lacks the N-terminus and accelerates the self-inactivating Kv channels. Interestingly, Kvb subunits have been proposed to act as sensors of lipid oxidation by reducing oxidized phospholipids generated during OS. These oxidized phospholipids can alter the inactivation properties of Kv channels, suggesting that the b-subunits modulate the detection of metabolic changes during oxidative stress [110][220][221].
Furthermore, both Kvb1 and Kvb2 exhibit aldo-keto reductase activity, functioning as redox enzymes. They can convert aldehydes to alcohols using NADPH as a cofactor. The cofactor is bound to the b-subunits, and its oxidation enhances the current flow through Kv1 channels. The oxidation of the cofactor is voltage dependent. Thus, the Kv1-Kvb complex enables the translation of the cellular redox state into changes in cellular excitability, effectively conveying the impact of oxidative conditions on cellular function [222].
Voltage-gated Kv7 (KCNQ family) potassium channels are present in numerous neuronal populations and have a crucial function in controlling membrane potential. They achieve this by producing a hyperpolarizing potassium current, which effectively reduces cellular excitability representing an important pharmacological target for hyperexcitability disorders [223][224]. Research has revealed that certain members of the Kv7 family (Kv7.2, Kv7.3, and Kv7.5) can combine to form heterotetramers responsible for carrying the ‘M’ current. This stabilizing potassium current is sensitive to inhibition by muscarinic receptor stimulation in neurons [225].
Recently, Peiris et al. identified Kv7 channels in enteric nervous system neurons along the gastrointestinal tract in humans and mice, including afferent fibers innervating the distal colon. In particular, activation of neuronal Kv7.3 channels has been shown to suppress both mechanically and chemically induced sensory responses [226]. Although, the impact of Kv7 channel activation on epithelial electrolyte transport remains unclear and requires further investigation.
According to the results of Nickerson et al., activation of Kv7 channels leads to a significant reduction in Cl− secretion by the mouse distal colon epithelium. This effect is probably attributed to the Kv7 channels present in the enteric neurons of the submucosal plexus, which decrease neuronal excitability and consequently inhibit stimulatory signals reaching the epithelial layer [227]. All these insights have promising clinical implications for the potential development of therapeutic interventions to mitigate CNS hyperexcitability in gastrointestinal disorders such as IBD and IBS.
ROS generated by oxidative stress are known to induce increased KCNQ currents in neurons [228]. This is mainly due to the methylation of protein arginine, which constitutes a post-translational modification that enhances the function of the KCNQ/M channel.
A critical characteristic of M-channels is their reliance on membrane phosphatidylinositol-4,5-bisphosphate (PIP2) to be able to open [229]. These channels contain numerous arginine residues within their PIP2-binding domain, and these residues can be subject to methylation by protein arginine methyltransferases (Prmts). Prmts are enzymes that catalyze the transfer of a methyl group to arginine residues and are highly expressed in the CNS [230]. The observation that decreased methylation lowers PIP2 affinity suggests that methylation of arginine residues enhances the channel-PIP2 interaction. The augmentation of KCNQ currents in response to oxidative stress has been linked to a significant reduction in apoptosis activation, which may help prevent neuronal death [231].
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 26, 757–772.
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969.
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215, 213–219.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19.
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049.
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763.
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472.
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 4535194.
- Ponnampalam, E.N.; Kiani, A.; Santhiravel, S.; Holman, B.W.B.; Lauridsen, C.; Dunshea, F.R. The Importance of Dietary Antioxidants on Oxidative Stress, Meat and Milk Production, and Their Preservative Aspects in Farm Animals: Antioxidant Action, Animal Health, and Product Quality-Invited Review. Animals 2022, 12, 3279.
- Udenigwe, C.C.; Aluko, R.E. Food protein-derived bioactive peptides: Production, processing, and potential health benefits. J. Food Sci. 2012, 1, R11–R24.
- Pepe, G.; Basilicata, M.G.; Carrizzo, A.; Adesso, S.; Ostacolo, C.; Sala, M.; Sommella, E.; Ruocco, M.; Cascioferro, S.; Ambrosio, M.; et al. β-Lactoglobulin Heptapeptide Reduces Oxidative Stress in Intestinal Epithelial Cells and Angiotensin II-Induced Vasoconstriction on Mouse Mesenteric Arteries by Induction of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Translocation. Oxidative Med. Cell. Longev. 2019, 2019, 1616239.
- Basilicata, M.G.; Pepe, G.; Rapa, S.F.; Merciai, F.; Ostacolo, C.; Manfra, M.; Di Sarno, V.; Autore, G.; De Vita, D.; Marzocco, S.; et al. Anti-Inflammatory and Antioxidant Properties of Dehydrated Potato-Derived Bioactive Compounds in Intestinal Cells. Int. J. Mol. Sci. 2019, 20, 6087.
- Khan, I.; Samson, S.E.; Grover, A.K. Antioxidant Supplements and Gastrointestinal Diseases: A Critical Appraisal. Med. Princ. Pract. 2017, 26, 201–217.
- Herrero de la Parte, B.; Rodeño-Casado, M.; Iturrizaga Correcher, S.; Mar Medina, C.; García-Alonso, I. Curcumin Reduces Colorectal Cancer Cell Proliferation and Migration and Slows In Vivo Growth of Liver Metastases in Rats. Biomedicines 2021, 9, 1183.
- Kumar, P.; Singh, A.; Kumar, A.; Kumar, R.; Pal, R.; Sachan, A.K.; Dixit, R.K.; Nath, R. Effect of Curcumin and Coenzyme Q10 Alone and in Combination on Learning and Memory in an Animal Model of Alzheimer’s Disease. Biomedicines 2023, 11, 1422.
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; et al. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009.
- Marino, P.; Pepe, G.; Basilicata, M.G.; Vestuto, V.; Marzocco, S.; Autore, G.; Procino, A.; Gomez-Monterrey, I.M.; Manfra, M.; Campiglia, P. Potential Role of Natural Antioxidant Products in Oncological Diseases. Antioxidants 2023, 12, 704.
- Quagliariello, V.; Basilicata, M.G.; Pepe, G.; De Anseris, R.; Di Mauro, A.; Scognamiglio, G.; Palma, G.; Vestuto, V.; Buccolo, S.; Luciano, A.; et al. Combination of Spirulina platensis, Ganoderma lucidum and Moringa oleiferaImproves Cardiac Functions and Reduces Pro-Inflammatory Biomarkers in Preclinical Models of Short-Term Doxorubicin-Mediated Cardiotoxicity: New Frontiers in Cardioncology? J. Cardiovasc. Dev. Dis. 2022, 9, 423.
- Dziąbowska-Grabias, K.; Sztanke, M.; Zając, P.; Celejewski, M.; Kurek, K.; Szkutnicki, S.; Korga, P.; Bulikowski, W.; Sztanke, K. Antioxidant Therapy in Inflammatory Bowel Diseases. Antioxidants 2021, 10, 412.
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189.
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201.
- Yoshida, N. Inflammation and oxidative stress in gastroesophageal reflux disease. J. Clin. Biochem. Nutr. 2007, 40, 13–23.
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24.
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40.
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438.
- Halliwell, B.; Chirico, S. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S.
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940.
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 69–76.
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289.
- Sohal, R.S. Role of oxidative stress and protein oxidation in the aging process. Free Radic. Biol. Med. 2002, 33, 37–44.
- Stadtman, E.R. Protein oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258.
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003, 25, 207–218.
- Collins, A.R. Oxidative DNA damage, antioxidants, and cancer. Bioessays 1999, 21, 238–246.
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214.
- Chiorcea-Paquim, A.M. 8-oxoguanine and 8-oxodeoxyguanosine Biomarkers of Oxidative DNA Damage: A Review on HPLC-ECD Determination. Molecules 2022, 27, 1620.
- Pereira, C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative Stress and DNA Damage: Implications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417.
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028.
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484.
- Swindle, E.J.; Metcalfe, D.D. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol. Rev. 2007, 217, 186–205.
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and Aging. Cell 2005, 120, 483–495.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
- Kulkarni, A.C.; Kuppusamy, P.; Parinandi, N. Oxygen, the lead actor in the pathophysio- logic drama: Enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid. Redox Signal. 2007, 9, 1717–1730.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Geiszt, M.; Leto, T.L. The Nox family of NAD(P)H oxidases: Host defense and beyond. J. Biol. Chem. 2004, 279, 51715–51718.
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454.
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156.
- Cerqua, I.; Musella, S.; Peltner, L.K.; D’Avino, D.; Di Sarno, V.; Granato, E.; Vestuto, V.; Di Matteo, R.; Pace, S.; Ciaglia, T.; et al. Discovery and Optimization of Indoline-Based Compounds as Dual 5-LOX/sEH Inhibitors: In Vitro and In Vivo Anti-Inflammatory Characterization. J. Med. Chem. 2022, 65, 14456–14480.
- Zhu, H.; Yang, L.; Zhou, B.; Yu, R.; Tang, N.; Wang, B. Myeloperoxidase G-463A polymorphism and the risk of gastric cancer: A case-control study. Carcinogenesis 2006, 27, 2491–2496.
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685.
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858.
- Kaneko, K.; Akuta, T.; Sawa, T.; Kim, H.W.; Fujii, S.; Okamoto, T.; Nakayama, H.; Ohigashi, H.; Murakami, A.; Akaike, T. Mutagenicity of 8-nitroguanosine, a product of nitrative nucleoside modification by reactive nitrogen oxides, in mammalian cells. Cancer Lett. 2008, 262, 239–247.
- Muñoz, M.; Sánchez, A.; Pilar Martínez, M.; Benedito, S.; López-Oliva, M.E.; García-Sacristán, A.; Hernández, M.; Prieto, D. COX-2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic. Biol. Med. 2015, 84, 77–90.
- Marnett, L.J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290.
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393.
- Kruidenier, L.; Kuiper, I.; Van Duijn, W.; Marklund, S.L.; Van Hogezand, R.A.; Lamers, C.B.; Verspaget, H.W. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J. Pathol. 2003, 201, 7–16.
- Janssen, A.M.; Bosman, C.B.; Van Duijn, W.; Oostendorp-van de Ruit, M.M.; Kubben, F.J.; Griffioen, G.; Lamers, C.B.; van Krieken, J.H.; van de Velde, C.J.; Verspaget, H.W. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res. 2000, 6, 3183–3192.
- Komatsu, H.; Okayasu, I.; Mitomi, H.; Imai, H.; Nakagawa, Y.; Obata, F. Immunohistochemical detection of human gastrointestinal glutathione peroxidase in normal tissues and cultured cells with novel mouse monoclonal antibodies. J. Histochem. Cytochem. 2001, 49, 759–766.
- Guan, G.; Lan, S. Implications of Antioxidant Systems in Inflammatory Bowel Disease. BioMed Res. Int. 2018, 2018, 1290179.
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236.
- Pérez, S.; Taléns-Visconti, R.; Rius-Pérez, S.; Finamor, I.; Sastre, J. Redox signaling in the gastrointestinal tract. Free Radic. Biol. Med. 2017, 104, 75–103.
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090.
- Hong, J.; Li, D.; Cao, W. Rho Kinase ROCK2 Mediates Acid-Induced NADPH Oxidase NOX5-S Expression in Human Esophageal Adenocarcinoma Cells. PLoS ONE 2016, 11, e0149735.
- Li, D.; Cao, W. Role of intracellular calcium and NADPH oxidase NOX5-S in acid-induced DNA damage in Barrett’s cells and Barrett’s esophageal adenocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G863–G872.
- Zhang, Q.B.; Nakashabendi, I.M.; Mokhashi, M.S.; Dawodu, J.B.; Gemmell, C.G.; Russell, R.I. Association of cytotoxin production and neutrophil activation by strains of Helicobacter pylori isolated from patients with peptic ulceration and chronic gastritis. Gut 1996, 38, 841–845.
- Chaturvedi, R.; Asim, M.; Barry, D.P.; Frye, J.W.; Casero, R.A., Jr.; Wilson, K.T. Spermine oxidase is a regulator of macrophage host response to Helicobacter pylori: Enhancement of antimicrobial nitric oxide generation by depletion of spermine. Amino Acids 2014, 46, 531–542.
- Colgan, S.P.; Taylor, C.T. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287.
- Iborra, M.; Moret, I.; Rausell, F.; Bastida, G.; Aguas, M.; Cerrillo, E.; Nos, P.; Beltran, B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem. Soc. Trans. 2011, 39, 1102–1106.
- Singer, I.I.; Kawka, D.W.; Scott, S.; Weidner, J.R.; Mumford, R.A.; Riehl, T.E.; Stenson, W.F. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 1996, 111, 871–885.
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Hypochlorous Acid Chemistry in Mammalian Cells—Influence on Infection and Role in Various Pathologies. Int. J. Mol. Sci. 2022, 23, 10735.
- Hendrickson, B.A.; Gokhale, R.; Cho, J.H. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 2002, 15, 79–94.
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86.
- Laurindo, L.F.; Santos, A.R.O.D.; Carvalho, A.C.A.; Bechara, M.D.; Guiguer, E.L.; Goulart, R.A.; Vargas Sinatora, R.; Araújo, A.C.; Barbalho, S.M. Phytochemicals and Regulation of NF-kB in Inflammatory Bowel Diseases: An Overview of In Vitro and In Vivo Effects. Metabolites 2023, 13, 96.
- Coskun, M.; Olsen, J.; Seidelin, J.B.; Nielsen, O.H. MAP kinases in inflammatory bowel disease. Clin. Chim. Acta 2011, 412, 513–520.
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611.
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372.
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199.
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384.
- Eugene, S.P.; Reddy, V.S.; Trinath, J. Endoplasmic Reticulum Stress and Intestinal Inflammation: A Perilous Union. Front. Immunol. 2020, 11, 543022.
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276.
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264.
- Qayyum, N.; Haseeb, M.; Kim, M.S.; Choi, S. Role of Thioredoxin-Interacting Protein in Diseases and Its Therapeutic Outlook. Int. J. Mol. Sci. 2021, 22, 2754.
- Liu, Z.; Leng, W.; Zhang, J.; Zhang, G.; Liu, D.; Zhao, Z.; Chen, F.; Shi, Y.; Hao, Y.; Lv, J.; et al. miR-146a-5p/TXNIP axis attenuates intestinal ischemia-reperfusion injury by inhibiting autophagy via the PRKAA/mTOR signaling pathway. Biochem. Pharmacol. 2022, 197, 114839.
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium. 2016, 60, 108–114.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Ramírez, A.; Vázquez-Sánchez, A.Y.; Carrión-Robalino, N.; Camacho, J. Ion Channels and Oxidative Stress as a Potential Link for the Diagnosis or Treatment of Liver Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 3928714.
- Annunziato, L.; Pannaccione, A.; Cataldi, M.; Secondo, A.; Castaldo, P.; Di Renzo, G.; Taglialatela, M. Modulation of ion channels by reactive oxygen and nitrogen species: A pathophysiological role in brain aging? Neurobiol. Aging 2002, 23, 819–834.
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051.
- O’Grady, S.M. Oxidative stress, autophagy and airway ion transport. Am. J. Physiol. Cell Physiol. 2019, 316, C16–C32.
- Beyder, A.; Farrugia, G. Ion channelopathies in functional GI disorders. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G581–G586.
- Akbarali, H.I.; Hawkins, E.G.; Ross, G.R.; Kang, M. Ion channel remodeling in gastrointestinal inflammation. Neurogastroenterol. Motil. 2010, 22, 1045–1055.
- Deng, Z.; Zhao, Y.; Ma, Z.; Zhang, M.; Wang, H.; Yi, Z.; Tuo, B.; Li, T.; Liu, X. Pathophysiological role of ion channels and transporters in gastrointestinal mucosal diseases. Cell. Mol. Life Sci. 2021, 78, 8109–8125.
- Hill-Eubanks, D.C.; Werner, M.E.; Heppner, T.J.; Nelson, M.T. Calcium signaling in smooth muscle. Cold Spring Harb. Perspect. Biol. 2011, 3, a004549.
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947.
- Drumm, B.T.; Hwang, S.J.; Baker, S.A.; Ward, S.M.; Sanders, K.M. Ca2+ signalling behaviours of intramuscular interstitial cells of Cajal in the murine colon. J. Physiol. 2019, 597, 3587–3617.
- Chen, H.; Ordög, T.; Chen, J.; Young, D.L.; Bardsley, M.R.; Redelman, D.; Ward, S.M.; Sanders, K.M. Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol. Genom. 2007, 31, 492–509.
- Terlau, H.; Stühmer, W. Structure and function of voltage-gated ion channels. Naturwissenschaft 1998, 85, 437–444.
- Beyder, A.; Farrugia, G. Targeting ion channels for the treatment of gastrointestinal motility disorders. Therap. Adv. Gastroenterol. 2012, 5, 5–21.
- Todorovic, S.M.; Jevtovic-Todorovic, V. Redox regulation of neuronal voltage-gated calcium channels. Antioxid. Redox Signal. 2014, 21, 880–891.
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555.
- Shi, X.Z.; Sarna, S.K. Impairment of Ca2+ mobilization in circular muscle cells of the inflamed colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G234–G242.
- Liu, X.; Rusch, N.J.; Striessnig, J.; Sarna, S.K. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology 2001, 120, 480–489.
- Ross, G.R.; Kang, M.; Shirwany, N.; Malykhina, A.P.; Drozd, M.; Akbarali, H.I. Nitrotyrosylation of Ca2+ channels prevents c-Src kinase regulation of colonic smooth muscle contractility in experimental colitis. J. Pharmacol. Exp. Ther. 2007, 322, 948–956.
- Ross, G.R.; Kang, M.; Akbarali, H.I. Colonic inflammation alters Src kinase-dependent gating properties of single Ca2+ channels via tyrosine nitration. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G976–G984.
- Wheeler, D.G.; Barrett, C.F.; Groth, R.D.; Safa, P.; Tsien, R.W. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 2008, 183, 849–863.
- Kang, M.; Ross, G.R.; Akbarali, H.I. The effect of tyrosine nitration of L-type Ca2+ channels on excitation-transcription coupling in colonic inflammation. Br. J. Pharmacol. 2010, 159, 1226–1235.
- Akbarali, H.I. Oxidative Stress and Ion Channels. In Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014.
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacology 2014, 66, 676–814.
- Ciaglia, T.; Vestuto, V.; Bertamino, A.; González-Muñiz, R.; Gómez-Monterrey, I. On the modulation of TRPM channels: Current perspectives and anticancer therapeutic implications. Front. Oncol. 2023, 12, 1065935.
- Daisuke, K.; Nozomi, O.; Yasuo, M. Redox Regulation of Transient Receptor Potential Channels. Antioxid. Redox Signal. 2014, 21, 971–986.
- Simon, F.; Varela, D.; Cabello-Verrugio, C. Oxidative stress-modulated TRPM ion channels in cell dysfunction and pathological conditions in humans. Cell. Signal. 2013, 25, 1614–1624.
- Holzer, P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacology 2011, 131, 142–170.
- Chen, Y.; Mu, J.; Zhu, M.; Mukherjee, A.; Zhang, H. Transient Receptor Potential Channels and Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 180.
- Du, Y.; Chen, J.; Shen, L.; Wang, B. TRP channels in inflammatory bowel disease: Potential therapeutic targets. Biochem. Pharmacol. 2022, 203, 115195.
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient Receptor Potential Melastatin 2 Is Required for Lipopolysaccharide-Induced Cytokine Production in Human Monocytes. J. Immunol. 2010, 184, 2386–2393.
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599.
- Perraud, A.L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium. 2003, 33, 519–531.
- Knowles, H.; Li, Y.; Perraud, A.L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013, 55, 241–248.
- Özdemir, Ü.S.; Nazıroğlu, M.; Şenol, N.; Ghazizadeh, V. Hypericum perforatum Attenuates Spinal Cord Injury-Induced Oxidative Stress and Apoptosis in the Dorsal Root Ganglion of Rats: Involvement of TRPM2 and TRPV1 Channels. Mol. Neurobiol. 2016, 53, 3540–3551.
- Buelow, B.; Song, Y.; Scharenberg, A.M. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583.
- Vestuto, V.; Di Sarno, V.; Musella, S.; Di Dona, G.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors? Int. J. Mol. Sci. 2023, 24, 185.
- Yamamoto, S.; Takahashi, N.; Mori, Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog. Biophys. Mol. Biol. 2010, 103, 18–27.
- Oda, S.; Uchida, K.; Wang, X.; Lee, J.; Shimada, Y.; Tominaga, M.; Kadowaki, M. TRPM2 contributes to antigen-stimulated Ca2+ influx in mucosal mast cells. Mol. Cell. Mech. Dis. 2013, 465, 1023–1030.
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747.
- Matsumoto, K.; Takagi, K.; Kato, A.; Ishibashi, T.; Mori, Y.; Tashima, K.; Mitsumoto, A.; Kato, S.; Horie, S. Role of transient receptor potential melastatin 2 (TRPM2) channels in visceral nociception and hypersensitivity. Exp. Neurol. 2016, 285, 41–50.
- Olah, M.E.; Jackson, M.F.; Li, H.; Perez, Y.; Sun, H.S.; Kiyonaka, S.; Mori, Y.; Tymianski, M.; MacDonald, J.F. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J. Physiol. 2009, 587, 965–979.
- Matsumoto, K.; Kawanaka, H.; Hori, M.; Kusamori, K.; Utsumi, D.; Tsukahara, T.; Amagase, K.; Horie, S.; Yamamoto, A.; Ozaki, H.; et al. Role of transient receptor potential melastatin 2 in surgical inflammation and dysmotility in a mouse model of postoperative ileus. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G104–G116.
- Uchida, K.; Tominaga, M. The role of TRPM2 in pancreatic β-cells and the development of diabetes. Cell Calcium 2014, 56, 332–339.
- Holzer, P. TRP channels in the digestive system. Curr. Pharm. Biotechnol. 2011, 12, 24–34.
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797.
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The Role of Transient Receptor Potential Vanilloid 1 in Common Diseases of the Digestive Tract and the Cardiovascular and Respiratory System. Front. Physiol. 2019, 10, 1064.
- Akbar, A.; Yiangou, Y.; Facer, P.; Walters, J.R.; Anand, P.; Ghosh, S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut 2008, 57, 923–929.
- Chuang, H.H.; Lin, S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 20097–20102.
- Schilling, T.; Eder, C. Importance of the non-selective cation channel TRPV1 for microglial reactive oxygen species generation. J. Neuroimmunol. 2009, 216, 118–121.
- Kishimoto, E.; Naito, Y.; Handa, O.; Okada, H.; Mizushima, K.; Hirai, Y.; Nakabe, N.; Uchiyama, K.; Ishikawa, T.; Takagi, T.; et al. Oxidative stress-induced posttranslational modification of TRPV1 expressed in esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G230–G238.
- Bhat, Y.M.; Bielefeldt, K. Capsaicin receptor (TRPV1) and non-erosive reflux disease. Eur. J. Gastroenterol. Hepatol. 2006, 18, 263–270.
- Matthews, P.J.; Aziz, Q.; Facer, P.; Davis, J.B.; Thompson, D.G.; Anand, P. Increased capsaicin receptor TRPV1 nerve fibres in the inflamed human oesophagus. Eur. J. Gastroenterol. Hepatol. 2004, 16, 897–902.
- Clarrett, D.M.; Hachem, C. Gastroesophageal Reflux Disease (GERD). MO Med. 2018, 115, 214–218.
- Cheng, L.; de la Monte, S.; Ma, J.; Hong, J.; Tong, M.; Cao, W.; Behar, J.; Biancani, P.; Harnett, K.M. HCl− Activated Neural and Epithelial Vanilloid Receptors (TRPV1) in Cat Esophageal Mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G135–G143.
- Banerjee, B.; Medda, B.K.; Lazarova, Z.; Bansal, N.; Shaker, R.; Sengupta, J.N. Effect of reflux-induced inflammation on transient receptor potential vanilloid one (TRPV1) expression in primary sensory neurons innervating the oesophagus of rats. Neurogastroenterol. Motil. 2007, 19, 681–691.
- Bautista, D.M. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282.
- Nozawa, K.; Kawabata-Shoda, E.; Doihara, H.; Kojima, R.; Okada, H.; Mochizuki, S.; Sano, Y.; Inamura, K.; Matsushime, H.; Koizumi, T.; et al. TRPA1 regulates gastrointestinal motility through serotonin release from enterochromaffin cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3408–3413.
- Nagata, K.; Duggan, A.; Kumar, G.; Garcia-Anoveros, J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 2005, 25, 4052–4061.
- Hinman, A.; Chuang, H.H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568.
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 20, 363–383.
- Liu, M.; Gao, L.; Zhang, N. Berberine reduces neuroglia activation and inflammation in streptozotocin-induced diabetic mice. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419866379.
- Zou, B.; Cao, C.; Fu, Y.; Pan, D.; Wang, W.; Kong, L. Berberine Alleviates Gastroesophageal Reflux-Induced Airway Hyperresponsiveness in a Transient Receptor Potential A1-Dependent Manner. Evid. Based Complement. Altern. Med. 2022, 2022, 7464147.
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Physiol. Behav. 2017, 176, 139–148.
- Herrmann, A.K.; Wüllner, V.; Moos, S.; Graf, J.; Chen, J.; Kieseier, B.; Kurschus, F.C.; Albrecht, P.; Vangheluwe, P.; Methner, A. Dimethyl fumarate alters intracellular Ca2+ handling in immune cells by redox-mediated pleiotropic effects. Free Radic. Biol. Med. 2019, 141, 338–347.
- Sawada, Y.; Hosokawa, H.; Matsumura, K.; Kobayashi, S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur. J. Neurosci. 2008, 27, 1131–1142.
- Liu, X.L.; Zhen, C.; Chen, Y.X.; Fan, X.R.; Wu, Z.Y.; Ma, F.C.; Rong, J.; Di, G.F.; Jiang, X.C. Inhibition of TRPA1 Attenuates Oxidative Stress-induced Damage After Traumatic Brain Injury via the ERK/AKT Signaling Pathway. Neuroscience 2022, 494, 51–68.
- Eid, S.R.; Crown, E.D.; Moore, E.L.; Liang, H.A.; Choong, K.C.; Dima, S.; Henze, D.A.; Kane, S.A.; Urban, M.O. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain 2008, 4, 48.
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558.
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 Ion Channels in Inflammatory Bowel Diseases: Potential Therapeutic Targets? Pharmaceuticals 2019, 12, 48.
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320.
- Olivier, A.K.; Gibson-Corley, K.N.; Meyerholz, D.K. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: Gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G459–G471.
- Trouvé, P.; Férec, C.; Génin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980.
- Kerbiriou, M.; Le Drévo, M.A.; Férec, C.; Trouvé, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta 2007, 1772, 1236–1249.
- Bresso, F.; Askling, J.; Astegiano, M.; Demarchi, B.; Sapone, N.; Rizzetto, M.; Gionchetti, P.; Lammers, K.M.; de Leone, A.; Riegler, G.; et al. Potential role for the common cystic fibrosis F508 mutation in Crohn’s disease. Inflamm. Bowel Dis. 2007, 13, 531–536.
- Collobert, M.; Bocher, O.; Le Nabec, A.; Génin, E.; Férec, C.; Moisan, S. CFTR Cooperative Cis-Regulatory Elements in Intestinal Cells. Int. J. Mol. Sci. 2021, 22, 2599.
- Chappe, V.; Hinkson, D.A.; Zhu, T.; Chang, X.B.; Riordan, J.R.; Hanrahan, J.W. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J. Physiol. 2003, 548, 39–52.
- Jia, Y.; Mathews, C.J.; Hanrahan, J.W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem. 1997, 272, 4978–4984.
- Moliteo, E.; Sciacca, M.; Palmeri, A.; Papale, M.; Manti, S.; Parisi, G.F.; Leonardi, S. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules 2022, 27, 5324.
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890.
- Bahmanyar, S.; Ekbom, A.; Askling, J.; Johannesson, M.; Montgomery, S.M. Cystic fibrosis gene mutations and gastrointestinal diseases. J. Cyst. Fibros. 2010, 9, 288–291.
- Zhang, Z.; Leir, S.-H.; Harris, A. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell Mol. Biol. 2015, 52, 387–396.
- Borcherding, D.C.; Siefert, M.E.; Lin, S.; Brewington, J.; Sadek, H.; Clancy, J.P.; Plafker, S.M.; Ziady, A.G. Clinically approved CFTR modulators rescue Nrf2 dysfunction in cystic fibrosis airway epithelia. J. Clin. 2019, 129, 3448–3463.
- Duranton, C.; Rubera, I.; Cougnon, M.; Melis, N.; Chargui, A.; Mograbi, B.; Tauc, M. CFTR is involved in the fine tuning of intracellular redox status: Physiological implications in cystic fibrosis. Am. J. Pathol. 2012, 181, 1367–1377.
- Lubelska, K.; Wiktorska, K.; Mielczarek, L.; Milczarek, M.; Zbroińska-Bregisz, I.; Chilmonczyk, Z. Sulforaphane regulates NFE2L2/Nrf2-dependent xenobiotic metabolism phase II and phase III enzymes differently in human colorectal cancer and untransformed epithelial colon cells. Nutr. Cancer 2016, 68, 1338–1348.
- Bhattacharya, R.; Blankenheim, Z.; Scott, P.M.; Cormier, R.T. CFTR and Gastrointestinal Cancers: An Update. J. Pers. Med. 2022, 12, 868.
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133.
- Zheng, W.; Kuhlicke, J.; Jäckel, K.; Eltzschig, H.K.; Singh, A.; Sjoblöm, M.; Riederer, B.; Weinhold, C.; Seidler, U.; Colgan, S.P. Hypoxia inducible factor-1 (HIF-l)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J. 2009, 23, 204–213.
- Swahn, H.; Harris, A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes 2019, 10, 235.
- Liu, X.; Chen, Y.; You, B.; Peng, Y.; Chen, Y.; Yang, Z.; Zhang, Y.; Chen, J. Molecular mechanism mediating enteric bacterial translocation after severe burn: The role of cystic fibrosis transmembrane conductance regulator. Burn 2021, 9, tkaa042.
- Hartzell, C.; Putzier, I.; Arreola, J. Calcium-activated chloride channels. Annu. Rev. Physiol. 2005, 167, 719–758.
- Kunzelmann, K.; Kongsuphol, P.; Chootip, K.; Toledo, C.; Martins, J.R.; Almaca, J.; Tian, Y.; Witzgall, R.; Ousingsawat, J.; Schreiber, R. Role of the Ca2+ -activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol. Chem. 2011, 392, 125–134.
- Kunzelmann, K.; Kongsuphol, P.; Aldehni, F.; Tian, Y.; Ousingsawat, J.; Warth, R.; Schreiber, R. Bestrophin and TMEM16-Ca(2+) activated Cl− channels with different functions. Cell Calcium. 2009, 46, 233–241.
- Barrett, K.E.; Keely, S.J. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 2000, 62, 535–572.
- Sanders, K.M.; Zhu, M.H.; Britton, F.; Koh, S.D.; Ward, S.M. Anoctamins and gastrointestinal smooth muscle excitability. Exp. Physiol. 2012, 97, 200–206.
- Jeulin, C.; Guadagnini, R.; Marano, F. Oxidant stress stimulates Ca2+-activated chloride channels in the apical activated membrane of cultured nonciliated human nasal epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L636–L646.
- Evans, S.R.; Thoreson, W.B.; Beck, C.L. Molecular and functional analyses of two new calcium-activated chloride channel family members from mouse eye and intestine. J. Biol. Chem. 2004, 279, 41792–41800.
- Ulmasov, B.; Bruno, J.; Woost, P.G.; Edwards, J.C. Tissue and subcellular distribution of CLIC1. BMC Cell Biol. 2007, 8, 8.
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772.
- Uretmen Kagiali, Z.C.; Saner, N.; Akdag, M.; Sanal, E.; Degirmenci, B.S.; Mollaoglu, G.; Ozlu, N. CLIC4 and CLIC1 bridge plasma membrane and cortical actin network for a successful cytokinesis. Life Sci. Alliance 2020, 3, e201900558.
- Argenzio, E.; Moolenaar, W.H. Emerging biological roles of Cl- intracellular channel proteins. J. Cell Sci. 2016, 129, 4165–4174.
- Tulk, B.M.; Kapadia, S.; Edwards, J.C. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am. J. Physiol. Cell Physiol. 2002, 282, C1103–C1112.
- Novarino, G.; Fabrizi, C.; Tonini, R.; Denti, M.A.; Malchiodi-Albedi, F.; Lauro, G.M.; Sacchetti, B.; Paradisi, S.; Ferroni, A.; Curmi, P.M.; et al. Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J. Neurosci. 2004, 24, 5322–5330.
- Chen, C.D.; Wang, C.S.; Huang, Y.H.; Chien, K.Y.; Liang, Y.; Chen, W.J.; Lin, K.H. Overexpression of CLIC1 in human gastric carcinoma and its clinicopathological significance. Proteomics 2007, 7, 155–167.
- Petrova, D.T.; Asif, A.R.; Armstrong, V.W.; Dimova, I.; Toshev, S.; Yaramov, N.; Oellerich, M.; Toncheva, D. Expression of chloride intracellular channel protein 1 (CLIC1) and tumor protein D52 (TPD52) as potential biomarkers for colorectal cancer. Clin. Biochem. 2008, 41, 1224–1236.
- Lu, D.; Le, Y.; Ding, J.; Dou, X.; Mao, W.; Zhu, J. CLIC1 Inhibition Protects Against Cellular Senescence and Endothelial Dysfunction Via the Nrf2/HO-1 Pathway. Cell Biochem. Biophys. 2021, 79, 239–252.
- Meng, M. CLIC1 facilitates cancer associated fibroblast activation and gastric cancer progression via integrins/NF-κB pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G836.
- Li, B.P.; Mao, Y.T.; Wang, Z.; Chen, Y.Y.; Wang, Y.; Zhai, C.Y.; Shi, B.; Liu, S.Y.; Liu, J.L.; Chen, J.Q. CLIC1 Promotes the Progression of Gastric Cancer by Regulating the MAPK/AKT Pathways. Cell. Physiol. Biochem. 2018, 46, 907–924.
- Wang, J.W.; Peng, S.Y.; Li, J.T.; Wang, Y.; Zhang, Z.P.; Cheng, Y.; Cheng, D.Q.; Weng, W.H.; Wu, X.S.; Fei, X.Z.; et al. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett. 2009, 281, 71–81.
- Pardo, L.A.; Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48.
- González, C.; Baez-Nieto, D.; Valencia, I.; Oyarzún, I.; Rojas, P.; Naranjo, D.; Latorre, R. K+ channels: Function-structural overview. Compr. Physiol. 2012, 2, 2087–2149.
- Coetzee, W.A.; Amarillo, Y.; Chiu, J.; Chow, A.; Lau, D.; McCormack, T.; Moreno, H.; Nadal, M.S.; Ozaita, A.; Pountney, D.; et al. Molecular diversity of K+ channels. Ann. N. Y. Acad. Sci. 1999, 868, 233–285.
- Pardo, L.A.; Contreras-Jurado, C.; Zientkowska, M.; Alves, F.; Stühmer, W. Role of voltage-gated potassium channels in cancer. J. Membr. Biol. 2005, 205, 115–124.
- Taglialatela, M.; Castaldo, P.; Iossa, S.; Pannaccione, A.; Fresi, A.; Ficker, E.; Annunziato, L. Regulation of the human ether-a-gogo related gene (HERG) K+ channels by reactive oxygen species. Proc. Natl. Acad. Sci. USA 1997, 94, 11698–11703.
- Su, Z.; Limberis, J.; Martin, R.L.; Xu, R.; Kolbe, K.; Heinemann, S.H.; Hoshi, T.; Cox, B.F.; Gintant, G.A. Functional consequences of methionine oxidation of hERG potassium channels. Biochem. Pharmacol. 2007, 74, 702–711.
- Zhou, M.; He, H.J.; Tanaka, O.; Suzuki, R.; Sekiguchi, M.; Yasuoka, Y.; Kawahara, K.; Itoh, H.; Abe, H. Localization of the sulphonylurea receptor subunits, SUR2A and SUR2B, in rat renal tubular epithelium. Tohoku J. Exp. Med. 2008, 214, 247–256.
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476.
- Miki, T.; Seino, S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell. Cardiol. 2005, 38, 917–925.
- Cui, Y.; Fan, Z. Mechanism of Kir6.2 channel inhibition by sulfhydryl modification: Pore block or allosteric gating? J. Physiol. 2002, 540, 731–741.
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.Y.; Wu, H.E.; Gemes, G. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: Action by direct S-nitrosylation. Mol. Pain 2009, 5, 12.
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96.
- Tonooka, N.; Oseid, E.; Zhou, H.; Harmon, J.S.; Robertson, R.P. Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clin. Transplant. 2007, 21, 767–772.
- Welsh, N.; Margulis, B.; Borg, L.A.; Wiklund, H.J.; Saldeen, J.; Flodström, M.; Mello, M.A.; Andersson, A.; Pipeleers, D.G.; Hellerström, C. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: Implications for the pathogenesis of insulin-dependent diabetes mellitus. Mol. Med. 1995, 1, 806–820.
- Gier, B.; Krippeit-Drews, P.; Sheiko, T.; Aguilar-Bryan, L.; Bryan, J.; Düfer, M.; Drews, G. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J. Clin. Investig. 2009, 119, 3246–3256.
- Fridlyand, L.E.; Philipson, L.H. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic beta-cells? Diabetes 2004, 53, 1942–1948.
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72.
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268.
- Kang, M.; Hashimoto, A.; Gade, A.; Akbarali, H.I. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G532–G539.
- Gade, A.R.; Kang, M.; Akbarali, H.I. Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol. Pharmacol. 2013, 83, 294–306.
- Jin, X.; Malykhina, A.P.; Lupu, F.; Akbarali, H.I. Altered gene expression and increased bursting activity of colonic smooth muscle ATP-sensitive K+ channels in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G274–G285.
- Heitzmann, D.; Warth, R. Physiology and pathophysiology of potassium channels in gastrointestinal epithelia. Physiol. Rev. 2008, 88, 1119–1182.
- Ottschytsch, N.; Raes, A.; Van Hoorick, D.; Snyders, D.J. Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc. Natl. Acad. Sci. USA 2002, 99, 7986–7991.
- Peri, R.; Wible, B.A.; Brown, A.M. Mutations in the Kv beta 2 binding site for NADPH and their effects on Kv1. 4. J. Biol. Chem. 2001, 276, 738–741.
- Pan, Y.; Weng, J.; Cao, Y.; Bhosle, R.C.; Zhou, M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J. Biol. Chem. 2008, 283, 8634–8642.
- Anderson, U.A.; Carson, C.; McCloskey, K.D. KCNQ currents and their contribution to resting membrane potential and the excitability of interstitial cells of Cajal from the guinea pig bladder. J. Urol. 2009, 182, 330–336.
- Musella, S.; Carotenuto, L.; Iraci, N.; Baroli, G.; Ciaglia, T.; Nappi, P.; Basilicata, M.G.; Salviati, E.; Barrese, V.; Vestuto, V.; et al. Beyond Retigabine: Design, Synthesis, and Pharmacological Characterization of a Potent and Chemically Stable Neuronal Kv7 Channel Activator with Anticonvulsant Activity. J. Med. Chem. 2022, 65, 11340–11364.
- Roche, J.P.; Westenbroek, R.; Sorom, A.J.; Hille, B.; Mackie, K.; Shapiro, M.S. Antibodies and a cysteine-modifying reagent show correspondence of M current in neurons to KCNQ2 and KCNQ3 K+ channels. Br. J. Pharmacol. 2002, 137, 1173–1186.
- Peiris, M.; Hockley, J.R.; Reed, D.E.; Smith, E.S.J.; Bulmer, D.C.; Blackshaw, L.A. Peripheral K(v)7 channels regulate visceral sensory function in mouse and human colon. Mol. Pain 2017, 13, 1744806917709371.
- Nickerson, A.J.; Rottgen, T.S.; Rajendran, V.M. Activation of KCNQ (KV7) K+ channels in enteric neurons inhibits epithelial Cl- secretion in mouse distal colon. Am. J. Physiol. Cell Physiol. 2021, 320, C1074–C1087.
- Gamper, N.; Zaika, O.; Li, Y.; Martin, P.; Hernandez, C.C.; Perez, M.R.; Wang, A.Y.; Jaffe, D.B.; Shapiro, M.S. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006, 25, 4996–5004.
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195.
- Kim, H.J.; Jeong, M.H.; Kim, K.R.; Jung, C.Y.; Lee, S.Y.; Kim, H.; Koh, J.; Vuong, T.A.; Jung, S.; Yang, H.; et al. Protein arginine methylation facilitates KCNQ channel-PIP2 interaction leading to seizure suppression. Elife 2016, 5, e17159.
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
31 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No