Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rebecca R Florke Gee | -- | 1938 | 2023-08-30 17:42:22 | | | |
| 2 | Camila Xu | Meta information modification | 1938 | 2023-08-31 04:20:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hoyos Sanchez, M.C.; Bayat, T.; Florke Gee, R.R.; Fon Tacer, K. PWS-Associated Genes, Their Imprinting, and Expression Pattern. Encyclopedia. Available online: https://encyclopedia.pub/entry/48656 (accessed on 25 July 2026).
Hoyos Sanchez MC, Bayat T, Florke Gee RR, Fon Tacer K. PWS-Associated Genes, Their Imprinting, and Expression Pattern. Encyclopedia. Available at: https://encyclopedia.pub/entry/48656. Accessed July 25, 2026.
Hoyos Sanchez, Maria Camila, Tara Bayat, Rebecca R. Florke Gee, Klementina Fon Tacer. "PWS-Associated Genes, Their Imprinting, and Expression Pattern" Encyclopedia, https://encyclopedia.pub/entry/48656 (accessed July 25, 2026).
Hoyos Sanchez, M.C., Bayat, T., Florke Gee, R.R., & Fon Tacer, K. (2023, August 30). PWS-Associated Genes, Their Imprinting, and Expression Pattern. In Encyclopedia. https://encyclopedia.pub/entry/48656
Hoyos Sanchez, Maria Camila, et al. "PWS-Associated Genes, Their Imprinting, and Expression Pattern." Encyclopedia. Web. 30 August, 2023.
Copy Citation
Prader–Willi syndrome (PWS, OMIM #176270) and Schaaf–Yang syndrome (SYS, OMIM #615547) are rare autosomal-dominant, imprinted genetic disorders caused by the loss of one or more normally active paternal genes in the chromosomal region of 15q11-q13, called the PWS region.
PWS
SYS
imprinting
hormone secretion
secretory granule
1. Introduction
Prader–Willi syndrome (PWS, OMIM #176270) and Schaaf–Yang syndrome (SYS, OMIM #615547) are rare autosomal-dominant, imprinted genetic disorders caused by the loss of one or more normally active paternal genes in the chromosomal region of 15q11-q13, called the PWS region. The two main molecular causes of PWS include paternal 15q11-q13 deletion, which is present in 65–75% of individuals with PWS, and maternal uniparental disomy in which both chromosome 15s are from the mother, present in 20–30% of cases. The remaining individuals possess defects in the genomic imprinting center (IC) or chromosome 15 translocations or inversions [1][2][3]. PWS affects approximately one in 15,000–20,000 individuals with around 400,000 cases worldwide [4], while the prevalence of SYS is <1/1,000,000 [5]. Major clinical features of PWS include intellectual and physical disabilities, obesity, maladaptive behaviors, and several endocrine dysfunctions, like growth retardation and hypogonadism [6]. SYS shares several symptoms with PWS yet is distinct. The underlying genetic cause of SYS is the disrupted expression of MAGEL2, one of the protein-coding genes within the PWS region on chromosome 15, due to mutations in the paternal copy [7][8][9].
2. PWS-Associated Genes, Their Imprinting, and Expression Pattern
2.1. MKRN3
MKRN3, a highly conserved PWS-associated gene sharing 82% similarity between humans and mice, is ubiquitously expressed in human and mouse tissues with high levels in the brain and testis [10][11][12]. The hypothalamic MKRN3 expression is high early in life and decreases before puberty initiation, an evolutionarily conserved pattern observed in mice, rats, and non-human primates [13]. MKRN3 belongs to the Makorin family of proteins that contain two to four C3H zinc finger domains, a unique Cys-His configuration, and a RING zinc finger domain that is critical for the activity of the RING subfamily of E3 ubiquitin ligases [12][14]. Paternal deletion or loss-of-function mutations in MKRN3 are thought to contribute to hypogonadism, infertility, and the rare cases of central precocious puberty (CPP) in PWS patients [10][15][16][17][18]. CPP is characterized by elevated expression and secretion of hypothalamic gonadotropin-releasing hormone (GnRH), resulting in the early development of secondary sexual characteristics [15][19]. Mkrn3 knockout mice phenocopy many symptomatic features of human CPP [20]. Although MKRN3’s function in regulating puberty initiation in mammals is not completely understood, CPP-associated mutations in MKRN3 result from reduced expression or loss of E3 ubiquitin ligase function. MKRN3 ubiquitinates MBD3 (methyl-DNA binding protein 3) and epigenetically silences GNRH1 [20]. Furthermore, MKRN3-mediated ubiquitination of poly(A)-binding proteins destabilizes GNRH1 mRNA in the hypothalamus [19]; thus, MKRN3 regulates GnRH on the transcriptional and translational level. MKRN3 also inhibits puberty onset through interaction with other proteins, including IGF2BP1 and NPTX-1 [21][22][23]. Additionally, an in vitro luciferase assay showed that MKRN3 inhibited the expression of kisspeptin and neurokinin B, neuropeptides that stimulate GnRH secretion [13], though only neurokinin B protein levels were increased in Mkrn3 knockout mice [21]. In short, MKRN3 is a neuroendocrine inhibitor upstream of GnRH, and MKRN3 loss-of-function mutations are the main genetic cause of CPP, including in PWS patients [19][20][24][25].
2.2. MAGEL2
MAGEL2 is a member of the melanoma-antigen (MAGE) gene family, which expanded from one gene in lower eukaryotes to more than 40 genes in eutherian mammals [26]. MAGEL2 encodes for a regulator protein of an E3 ubiquitin ligase and affects the retromer-dependent endosomal protein recycling [7][27][28][29]. MAGEL2 is highly expressed in the central nervous system, especially in the hypothalamus and placenta [7][30]. Magel2-null mice recapitulate many PWS features, like poor sucking and obesity [31][32]. The loss of MAGEL2 leads to decreased neuropeptide and hormone production and impaired hypothalamic secretion. Further details about MAGEL2's function can be found here: [33].
2.3. NECDIN
Necdin (encoded by NDN) is another member of the MAGE family within the PWS region. NDN is highly expressed in certain regions of the brain, such as the locus coeruleus and hypothalamus and placenta [26][34][35][36]. In particular, NDN is highly expressed in GnRH neurons in the mature hypothalamus [37]. Necdin plays an integral role in neuronal differentiation [35], so Necdin deficiency leads to widespread nervous system abnormalities [38]. Deletion of Ndn in mice recapitulates several PWS symptoms, including neonatal mortality, altered pain threshold, hypogonadism, and sensory–motor defects [38][39][40], and significantly reduces the quantity of hypothalamic GnRH neurons [36][41]. Muscatelli et al. [42][43] reported a significant reduction in oxytocin-expressing neurons in the lateral parts of the paraventricular hypothalamic nucleus of Ndn-deficient mice. Necdin-deficient mice also exhibit disturbed migration of serotonin neuronal precursors and increased serotonin transporter activity that causes apnea, making Ndn knockout mice the only model that reproduces the respiratory challenges of the PWS [40][43][44]. Further, Necdin and Magel2 together were shown to control leptin receptor sorting and degradation through a ubiquitin-dependent pathway, including E3 ubiquitin ligase Rnf41, deubiquitinase Usp8, and protein Stam1, contributing to obesity in PWS [32]. More recently, Necdin was reported to regulate the stability of BMAL1, one of the core transcription regulators of the circadian rhythm, potentially contributing to the disturbed circadian rhythm observed in patients [34].
2.4. NPAP1
The imprinted NPAP1 is a primate-specific gene encoding a nuclear pore complex-associated protein from a POM121-related family of retrogenes with testis-specific expression, a unique pattern among the PWS genes [45][46][47]. Interestingly, NPAP1 is the only PWS gene not conserved in rodents. Rather, the PWS syntenic region on chromosomes 7 and 1 in mice and rats, respectively, contains another coding gene, Frat3, that is potentially involved in WNT signaling during embryonic development [48].
2.5. SNURF/SNRPN
SNURF/SNRPN (SNRPN upstream reading frame (SNURF)/small nuclear ribonucleoprotein polypeptide N (SNRPN)) is a complex gene locus belonging to the SNRPN SmB/SmN family. The protein plays a role in pre-mRNA processing, tissue-specific alternative splicing events, and transcript production [4]. The SNURF/SNRPN gene is a bicistronic transcript that encodes two proteins and also contains the snoRNA genes [49][50][51][52]. Chromosomal deletions that affect the SNRPN upstream exons and the imprinting center (IC) cause PWS by impairing the allele-specific expression of genes normally subject to the imprinting control [53]. It has also been reported that even a single small deletion or single-nucleotide variant involving SNURF/SNRPN causes major symptoms of PWS including hypotonia, dysmorphic features, intellectual disability, and obesity [54][55].
2.6. SNORD116
The non-coding RNA molecule SNORD116 is highly expressed in the brain [56], and clinical evidence from rare patients with SNORD116 deletions or translocations indicates that the SNORD116 cluster is crucial for most of the PWS phenotypes [10][57][58][59][60][61][62]. In mice, global or selective deletion of Snord116 from hypothalamic neurons causes low birth weight, increased weight gain in early adulthood, increased energy expenditure, and hyperphagia [63]. One group reported that Snord116 paternal knockout (Snord116m+/p−) mice also had reduced transcript levels of the prohormone convertase PC1 (encoded by Pcsk1), impairing prohormone processing and possibly causing the major neuroendocrine features of PWS [64]. Chen et al. [65] also observed a reduction in PC1 protein levels in pancreatic islets from Snord116m+/p− mice, although a follow-up study found no differences in the hypothalamic Pcsk1 transcript levels in Snord116m+/p− mice [66].
Besides PWS and SYS, Angelman syndrome (AS, OMIM#105830) is another imprinting disorder that is caused by genetic variation in the same region of chromosome 15. Ataxia, happy demeanor, and sleeplessness are some of the symptoms observed in individuals with AS [67][68]. In AS, the maternal copy of the genes in 15q11-q13 is missing, while the paternal copy is inactivated in PWS and SYS [7][68]. The PWS region is maternally imprinted, and genes must be expressed from the paternal chromosome. In contrast, the adjacent AS region is paternally imprinted, and encoded genes must be expressed from the maternal chromosome.
2.7. Genomic Imprinting
Genomic imprinting in PWS and AS causes a monoallelic expression of genes and is regulated by a bipartite IC, composed of the PWS-IC and AS-IC, that establishes local imprinting regulation of multiple genes within the 15q11-q13 region [69][70]. The PWS-IC comprises a CpG island and is associated with the 5′ flanking region, the first exon and 5′ end of the first intron of SNRPN [71]. The AS-IC is located 35 kb upstream of the SNRPN promoter [72]. The PWS-IC is differentially methylated on the maternal allele, with the paternal allele remaining in an open, unmethylated state [51][70][73][74][75]. Interestingly, deletion of AS-IC on the maternal allele also leads to the biallelic unmethylation of neighboring PWS-IC, suggesting that AS-IC contributes to establishing the methylation state and closed chromatin structure of PWS-IC [70][76]. Paternally inherited deletion of the PWS-IC results in loss of expression of MAGEL2 and other PWS genes [3][71]. For example, upon deletion of murine paternal PWS-IC, Brant et al. [77] noted a two-fold increase in the methylation level of CpG sites at differentially methylated regions (DMRs) of paternally expressed PWS genes, including Magel2, while deletion of maternal PWS-IC resulted in no methylation changes.
Although there are contradictory findings regarding when the maternal PWS-IC becomes methylated, in oocytes or post-fertilization, most results suggest that methylation occurs after the blastula stage [78][79][80][81]. A proposed model for PWS imprinting regulation suggests that methyl groups are removed from PWS-IC during both spermatogenesis and oogenesis, and then AS-IC interacts with PWS-IC to facilitate de novo methylation of the maternal PWS-IC after fertilization [71]. After fertilization and establishment of methylation of the maternal PWS-IC, the unmethylated paternal PWS-IC functions as a promoter for the SNRPN transcription unit and acts at long distances to activate transcription of MAGEL2, amongst others [71]. However, the exact mechanism and proteins involved in this intricate process are unclear.
2.8. Expression Pattern
In addition to the epigenetic regulation of imprinted genes in the PWS region, the distinct tissue expression patterns of these genes clearly suggest specific transcriptional regulation; however, the details of potential transcription factors involved are almost completely unknown. Interestingly, MAGEL2 and NDN are expressed at the highest levels in the brain (especially MAGEL2 in the hypothalamus), pituitary, and placenta [30][82], tissues linked to the evolution of genomic imprinting in eutherian mammals. For instance, most of the imprinted genes (~230 genes known so far) are expressed in the placenta and several now have well-known roles in placental biology, fetal growth, and homeostasis of pregnancy [83][84]. The functional convergence of many imprinted genes on the placenta is in line with the hypothesis that genomic imprinting evolved in mammals because of the conflicting interests of maternal and paternal genes in relation to the transfer of nutrients from the mother to her offspring [85][86]. Very little is known about the role of the PWS genes in the placenta and warrants future investigation.
Furthermore, genomic imprinting is increasingly appreciated for its role in the nervous system. Outside of the placenta, the brain is one of the adult tissues with the largest number of expressed imprinted genes [87][88]. In a recent tissue expression analysis on a single-cell level, imprinted genes were found over-represented in murine hypothalamic neurons [89]. These data support the previously suggested role of imprinted genes in the neuroendocrine hypothalamic regulation of diverse physiological functions [90]. Indeed, the first insights into the physiology of imprinted gene function were derived from studying neuroendocrine symptoms in PWS patients and animal models [91]. Through connections with the pituitary, the hypothalamus regulates hormone release in distal endocrine glands, including adrenals, thyroid, and gonads, to control key physiological processes, like stress response, growth, and reproduction. In addition, the hypothalamus makes neural connections via the autonomic nervous system and other pathways to regulate sleep, body temperature, and feeding [92], several of which are disturbed in PWS and SYS (Figure 1).
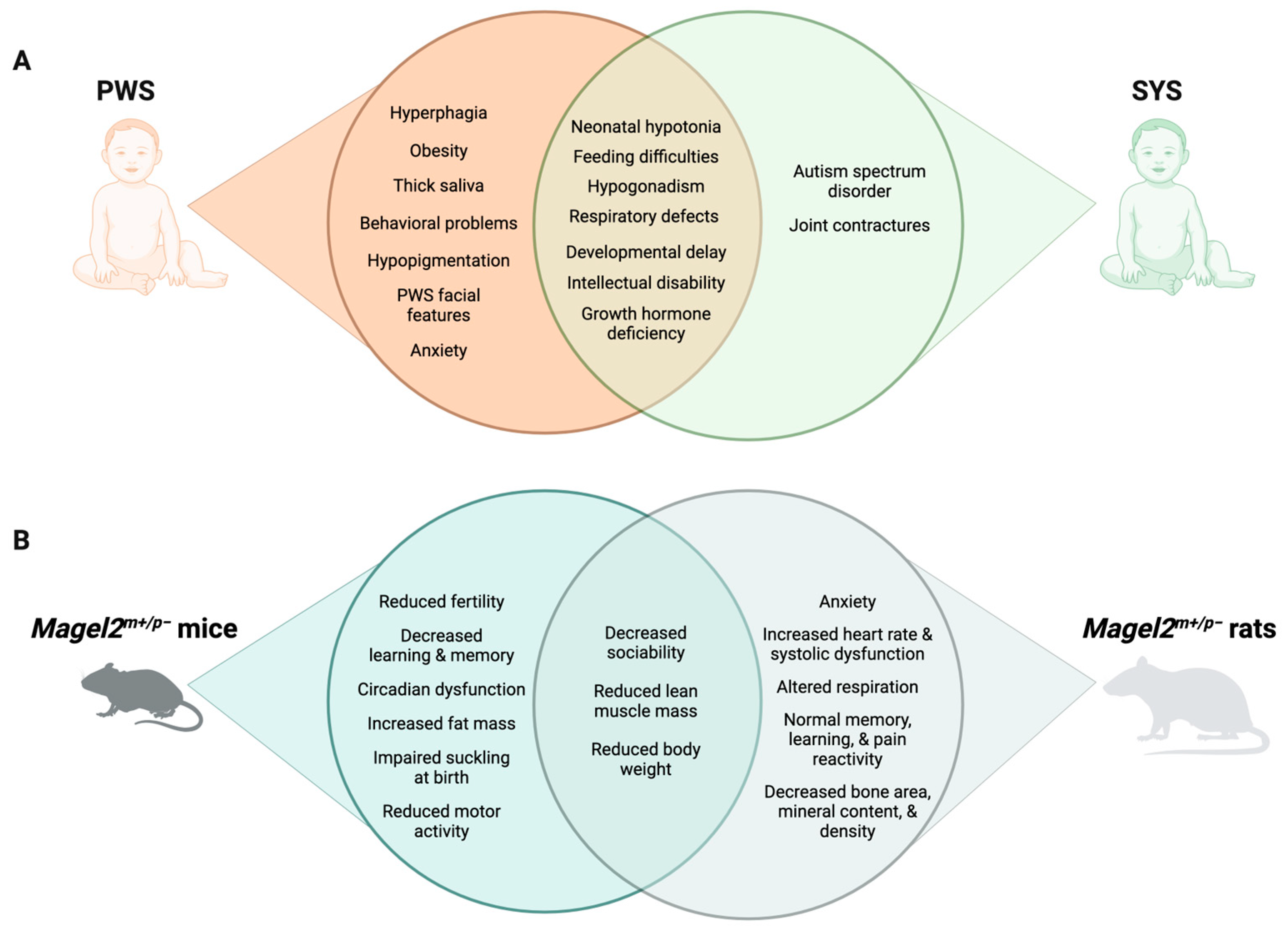
Figure 1. Comparison of PWS and SYS phenotypes in humans (A) and rodent models (B).
To summarize, mouse models and unique genetic backgrounds of patients revealed how single or multiple genes contribute to the development of PWS and SYS symptoms. The unique expression pattern of PWS genes (e.g., MAGEL2), their evolutionary appearance, and their molecular and cellular roles suggest that some of the PWS genes evolved in mammals as tissue-specific regulators of hypothalamic neuroendocrine function.
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26.
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153.
- Nicholls, R.D.; Knepper, J.L. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genom. Hum. Genet. 2001, 2, 153–175.
- Butler, M.G. Prader-Willi Syndrome and Chromosome 15q11.2 BP1-BP2 Region: A Review. Int. J. Mol. Sci. 2023, 24, 4271.
- Marbach, F.; Elgizouli, M.; Rech, M.; Beygo, J.; Erger, F.; Velmans, C.; Stumpel, C.; Stegmann, A.P.A.; Beck-Wodl, S.; Gillessen-Kaesbach, G.; et al. The adult phenotype of Schaaf-Yang syndrome. Orphanet J. Rare Dis. 2020, 15, 294.
- Driscoll, D.J.; Miller, J.L.; Cassidy, S.B. Prader-Willi Syndrome. In GeneReviews ((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Fon Tacer, K.; Potts, P.R. Cellular and disease functions of the Prader-Willi Syndrome gene MAGEL2. Biochem. J. 2017, 474, 2177–2190.
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408.
- Fountain, M.D.; Aten, E.; Cho, M.T.; Juusola, J.; Walkiewicz, M.A.; Ray, J.W.; Xia, F.; Yang, Y.; Graham, B.H.; Bacino, C.A.; et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 2017, 19, 45–52.
- Costa, R.A.; Ferreira, I.R.; Cintra, H.A.; Gomes, L.H.F.; Guida, L.D.C. Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome. Front. Endocrinol. 2019, 10, 864.
- Jong, M.T.; Gray, T.A.; Ji, Y.; Glenn, C.C.; Saitoh, S.; Driscoll, D.J.; Nicholls, R.D. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum. Mol. Genet. 1999, 8, 783–793.
- Jong, M.T.; Carey, A.H.; Caldwell, K.A.; Lau, M.H.; Handel, M.A.; Driscoll, D.J.; Stewart, C.L.; Rinchik, E.M.; Nicholls, R.D. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader-Willi syndrome genetic region. Hum. Mol. Genet. 1999, 8, 795–803.
- Abreu, A.P.; Toro, C.A.; Song, Y.B.; Navarro, V.M.; Bosch, M.A.; Eren, A.; Liang, J.N.; Carroll, R.S.; Latronico, A.C.; Ronnekleiv, O.K.; et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. J. Clin. Investig. 2020, 130, 4486–4500.
- Gray, T.A.; Hernandez, L.; Carey, A.H.; Schaldach, M.A.; Smithwick, M.J.; Rus, K.; Marshall Graves, J.A.; Stewart, C.L.; Nicholls, R.D. The ancient source of a distinct gene family encoding proteins featuring RING and C(3)H zinc-finger motifs with abundant expression in developing brain and nervous system. Genomics 2000, 66, 76–86.
- Meader, B.N.; Albano, A.; Sekizkardes, H.; Delaney, A. Heterozygous Deletions in MKRN3 Cause Central Precocious Puberty Without Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, 2732–2739.
- Macedo, D.B.; Abreu, A.P.; Reis, A.C.; Montenegro, L.R.; Dauber, A.; Beneduzzi, D.; Cukier, P.; Silveira, L.F.; Teles, M.G.; Carroll, R.S.; et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J. Clin. Endocrinol. Metab. 2014, 99, E1097–E1103.
- Ludwig, N.G.; Radaeli, R.F.; Silva, M.M.; Romero, C.M.; Carrilho, A.J.; Bessa, D.; Macedo, D.B.; Oliveira, M.L.; Latronico, A.C.; Mazzuco, T.L. A boy with Prader-Willi syndrome: Unmasking precocious puberty during growth hormone replacement therapy. Arch. Endocrinol. Metab. 2016, 60, 596–600.
- Lee, H.S.; Hwang, J.S. Central precocious puberty in a girl with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2013, 26, 1201–1204.
- Li, C.; Han, T.; Li, Q.; Zhang, M.; Guo, R.; Yang, Y.; Lu, W.; Li, Z.; Peng, C.; Wu, P.; et al. MKRN3-mediated ubiquitination of Poly(A)-binding proteins modulates the stability and translation of GNRH1 mRNA in mammalian puberty. Nucleic Acids Res. 2021, 49, 3796–3813.
- Li, C.; Lu, W.; Yang, L.; Li, Z.; Zhou, X.; Guo, R.; Wang, J.; Wu, Z.; Dong, Z.; Ning, G.; et al. MKRN3 regulates the epigenetic switch of mammalian puberty via ubiquitination of MBD3. Natl. Sci. Rev. 2020, 7, 671–685.
- Naule, L.; Mancini, A.; Pereira, S.A.; Gassaway, B.M.; Lydeard, J.R.; Magnotto, J.C.; Kim, H.K.; Liang, J.; Matos, C.; Gygi, S.P.; et al. MKRN3 inhibits puberty onset via interaction with IGF2BP1 and regulation of hypothalamic plasticity. JCI Insight 2023, 8, e164178.
- Liu, H.; Kong, X.; Chen, F. Mkrn3 functions as a novel ubiquitin E3 ligase to inhibit Nptx1 during puberty initiation. Oncotarget 2017, 8, 85102–85109.
- Yellapragada, V.; Liu, X.; Lund, C.; Kansakoski, J.; Pulli, K.; Vuoristo, S.; Lundin, K.; Tuuri, T.; Varjosalo, M.; Raivio, T. MKRN3 Interacts With Several Proteins Implicated in Puberty Timing but Does Not Influence GNRH1 Expression. Front. Endocrinol. 2019, 10, 48.
- Valadares, L.P.; Meireles, C.G.; De Toledo, I.P.; de Oliveira, R.S.; de Castro, L.C.G.; Abreu, A.P.; Carroll, R.S.; Latronico, A.C.; Kaiser, U.B.; Guerra, E.N.S.; et al. MKRN3 Mutations in Central Precocious Puberty: A Systematic Review and Meta-Analysis. J. Endocr. Soc. 2019, 3, 979–995.
- Abreu, A.P.; Dauber, A.; Macedo, D.B.; Noel, S.D.; Brito, V.N.; Gill, J.C.; Cukier, P.; Thompson, I.R.; Navarro, V.M.; Gagliardi, P.C.; et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 2013, 368, 2467–2475.
- Florke Gee, R.R.; Chen, H.; Lee, A.K.; Daly, C.A.; Wilander, B.A.; Fon Tacer, K.; Potts, P.R. Emerging roles of the MAGE protein family in stress response pathways. J. Biol. Chem. 2020, 295, 16121–16155.
- Doyle, J.M.; Gao, J.; Wang, J.; Yang, M.; Potts, P.R. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 2010, 39, 963–974.
- Hao, Y.H.; Fountain, M.D., Jr.; Fon Tacer, K.; Xia, F.; Bi, W.; Kang, S.H.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell 2015, 59, 956–969.
- Hao, Y.H.; Doyle, J.M.; Ramanathan, S.; Gomez, T.S.; Jia, D.; Xu, M.; Chen, Z.J.; Billadeau, D.D.; Rosen, M.K.; Potts, P.R. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell 2013, 152, 1051–1064.
- Fon Tacer, K.; Montoya, M.C.; Oatley, M.J.; Lord, T.; Oatley, J.M.; Klein, J.; Ravichandran, R.; Tillman, H.; Kim, M.; Connelly, J.P.; et al. MAGE cancer-testis antigens protect the mammalian germline under environmental stress. Sci. Adv. 2019, 5, eaav4832.
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905.
- Wijesuriya, T.M.; De Ceuninck, L.; Masschaele, D.; Sanderson, M.R.; Carias, K.V.; Tavernier, J.; Wevrick, R. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 2017, 26, 4215–4230.
- Maria Camila Hoyos Sanchez; Tara Bayat; Rebecca R. Florke Gee; Klementina Fon Tacer; Hormonal Imbalances in Prader–Willi and Schaaf–Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals. Int. J. Mol. Sci. 2023, 24, 13109.
- Lu, R.; Dong, Y.; Li, J.D. Necdin regulates BMAL1 stability and circadian clock through SGT1-HSP90 chaperone machinery. Nucleic Acids Res. 2020, 48, 7944–7957.
- Kuwajima, T.; Nishimura, I.; Yoshikawa, K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J. Neurosci. 2006, 26, 5383–5392.
- Miller, N.L.; Wevrick, R.; Mellon, P.L. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 2009, 18, 248–260.
- Napolitano, L.; Barone, B.; Morra, S.; Celentano, G.; La Rocca, R.; Capece, M.; Morgera, V.; Turco, C.; Caputo, V.F.; Spena, G.; et al. Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 1993.
- Wu, R.N.; Hung, W.C.; Chen, C.T.; Tsai, L.P.; Lai, W.S.; Min, M.Y.; Wong, S.B. Firing activity of locus coeruleus noradrenergic neurons decreases in necdin-deficient mice, an animal model of Prader-Willi syndrome. J. Neurodev. Disord. 2020, 12, 21.
- Bervini, S.; Herzog, H. Mouse models of Prader-Willi Syndrome: A systematic review. Front. Neuroendocr. 2013, 34, 107–119.
- Gerard, M.; Hernandez, L.; Wevrick, R.; Stewart, C.L. Disruption of the mouse necdin gene results in early post-natal lethality. Nat. Genet. 1999, 23, 199–202.
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752.
- Watrin, F.; Roeckel, N.; Lacroix, L.; Mignon, C.; Mattei, M.G.; Disteche, C.; Muscatelli, F. The mouse Necdin gene is expressed from the paternal allele only and lies in the 7C region of the mouse chromosome 7, a region of conserved synteny to the human Prader-Willi syndrome region. Eur. J. Hum. Genet. 1997, 5, 324–332.
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110.
- Matarazzo, V.; Caccialupi, L.; Schaller, F.; Shvarev, Y.; Kourdougli, N.; Bertoni, A.; Menuet, C.; Voituron, N.; Deneris, E.; Gaspar, P.; et al. Necdin shapes serotonergic development and SERT activity modulating breathing in a mouse model for Prader-Willi syndrome. Elife 2017, 6, e32640.
- Farber, C.; Gross, S.; Neesen, J.; Buiting, K.; Horsthemke, B. Identification of a testis-specific gene (C15orf2) in the Prader-Willi syndrome region on chromosome 15. Genomics 2000, 65, 174–183.
- Neumann, L.C.; Markaki, Y.; Mladenov, E.; Hoffmann, D.; Buiting, K.; Horsthemke, B. The imprinted NPAP1/C15orf2 gene in the Prader-Willi syndrome region encodes a nuclear pore complex associated protein. Hum. Mol. Genet. 2012, 21, 4038–4048.
- Neumann, L.C.; Feiner, N.; Meyer, A.; Buiting, K.; Horsthemke, B. The imprinted NPAP1 gene in the Prader-Willi syndrome region belongs to a POM121-related family of retrogenes. Genome Biol. Evol. 2014, 6, 344–351.
- Jonkers, J.; van Amerongen, R.; van der Valk, M.; Robanus-Maandag, E.; Molenaar, M.; Destree, O.; Berns, A. In vivo analysis of Frat1 deficiency suggests compensatory activity of Frat3. Mech. Dev. 1999, 88, 183–194.
- Kummerfeld, D.M.; Raabe, C.A.; Brosius, J.; Mo, D.; Skryabin, B.V.; Rozhdestvensky, T.S. A Comprehensive Review of Genetically Engineered Mouse Models for Prader-Willi Syndrome Research. Int. J. Mol. Sci. 2021, 22, 3613.
- Gray, T.A.; Saitoh, S.; Nicholls, R.D. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 5616–5621.
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 1996, 58, 335–346.
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135.
- Carias, K.V.; Wevrick, R. Preclinical Testing in Translational Animal Models of Prader-Willi Syndrome: Overview and Gap Analysis. Mol. Ther. Methods Clin. Dev. 2019, 13, 344–358.
- Cao, Y.; AlHumaidi, S.S.; Faqeih, E.A.; Pitel, B.A.; Lundquist, P.; Aypar, U. A novel deletion of SNURF/SNRPN exon 1 in a patient with Prader-Willi-like phenotype. Eur. J. Med. Genet. 2017, 60, 416–420.
- Huang, Y.; Grand, K.; Kimonis, V.; Butler, M.G.; Jain, S.; Huang, A.Y.; Martinez-Agosto, J.A.; Nelson, S.F.; Sanchez-Lara, P.A. Mosaic de novo SNRPN gene variant associated with Prader-Willi syndrome. J. Med. Genet. 2022, 59, 719–722.
- Basak, S.; Basak, A. Proteins and proteases of Prader-Willi syndrome: A comprehensive review and perspectives. Biosci. Rep. 2022, 42, BSR20220610.
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255.
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201.
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721.
- de Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265.
- Kim, S.J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290.
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700.
- Qi, Y.; Purtell, L.; Fu, M.; Lee, N.J.; Aepler, J.; Zhang, L.; Loh, K.; Enriquez, R.F.; Baldock, P.A.; Zolotukhin, S.; et al. Snord116 is critical in the regulation of food intake and body weight. Sci. Rep. 2016, 6, 18614.
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Investig. 2017, 127, 293–305.
- Chen, H.; Victor, A.K.; Klein, J.; Tacer, K.F.; Tai, D.J.; de Esch, C.; Nuttle, A.; Temirov, J.; Burnett, L.C.; Rosenbaum, M.; et al. Loss of MAGEL2 in Prader-Willi syndrome leads to decreased secretory granule and neuropeptide production. JCI Insight 2020, 5, e138576.
- Polex-Wolf, J.; Lam, B.Y.; Larder, R.; Tadross, J.; Rimmington, D.; Bosch, F.; Cenzano, V.J.; Ayuso, E.; Ma, M.K.; Rainbow, K.; et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J. Clin. Investig. 2018, 128, 960–969.
- Polvora-Brandao, D.; Joaquim, M.; Godinho, I.; Aprile, D.; Alvaro, A.R.; Onofre, I.; Raposo, A.C.; Pereira de Almeida, L.; Duarte, S.T.; da Rocha, S.T. Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet. 2018, 27, 3999–4011.
- Salminen, I.I.; Crespi, B.J.; Mokkonen, M. Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes. SAGE Open Med. 2019, 7, 2050312118823585.
- Barlow, D.P. Genomic imprinting: A mammalian epigenetic discovery model. Annu. Rev. Genet. 2011, 45, 379–403.
- Chung, M.S.; Langouet, M.; Chamberlain, S.J.; Carmichael, G.G. Prader-Willi syndrome: Reflections on seminal studies and future therapies. Open Biol. 2020, 10, 200195.
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A 2008, 146, 2041–2052.
- Buiting, K.; Lich, C.; Cottrell, S.; Barnicoat, A.; Horsthemke, B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum. Genet. 1999, 105, 665–666.
- Shemer, R.; Hershko, A.Y.; Perk, J.; Mostoslavsky, R.; Tsuberi, B.; Cedar, H.; Buiting, K.; Razin, A. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat. Genet. 2000, 26, 440–443.
- Saitoh, S.; Wada, T. Parent-of-Origin Specific Histone Acetylation and Reactivation of a Key Imprinted Gene Locus in Prader-Willi Syndrome. Am. J. Hum. Genet. 2000, 66, 1958–1962.
- Schweizer, J.; Zynger, D.; Francke, U. In vivo Nuclease Hypersensitivity Studies Reveal Multiple Sites of Parental Origin-Dependent Differential Chromatin Conformation in the 150 Kb SNRPN Transcription Unit. Hum. Mol. Genet. 1999, 8, 555–566.
- Perk, J.; Makedonski, K.; Lande, L.; Cedar, H.; Razin, A.; Shemer, R. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J. 2002, 21, 5807–5814.
- Brant, J.O.; Riva, A.; Resnick, J.L.; Yang, T.P. Influence of the Prader-Willi syndrome imprinting center on the DNA methylation landscape in the mouse brain. Epigenetics 2014, 9, 1540–1556.
- El-Maarri, O.; Buiting, K.; Peery, E.G.; Kroisel, P.M.; Balaban, B.; Wagner, K.; Urman, B.; Heyd, J.; Lich, C.; Brannan, C.I.; et al. Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat. Genet. 2001, 27, 341–344.
- Huntriss, J.; Hinkins, M.; Oliver, B.; Harris, S.E.; Beazley, J.C.; Rutherford, A.J.; Gosden, R.G.; Lanzendorf, S.E.; Picton, H.M. Expression of mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line, preimplantation embryos, and embryonic stem cells. Mol. Reprod. Dev. 2004, 67, 323–336.
- Kantor, B.; Kaufman, Y.; Makedonski, K.; Razin, A.; Shemer, R. Establishing the epigenetic status of the Prader–Willi/Angelman imprinting center in the gametes and embryo. Hum. Mol. Genet. 2004, 13, 2767–2779.
- DuBose, A.J.; Smith, E.Y.; Johnstone, K.A.; Resnick, J.L. Temporal and developmental requirements for the Prader-Willi imprinting center. Proc. Natl. Acad. Sci. USA 2012, 109, 3446–3450.
- Jiang, C.; Yang, Y.; Huang, C.; Whitelaw, B. Promoter characterization and functional association with placenta of porcine MAGEL2. Gene 2014, 547, 63–69.
- Renfree, M.B.; Hore, T.A.; Shaw, G.; Graves, J.A.; Pask, A.J. Evolution of genomic imprinting: Insights from marsupials and monotremes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 241–262.
- Plasschaert, R.N.; Bartolomei, M.S. Genomic imprinting in development, growth, behavior and stem cells. Development 2014, 141, 1805–1813.
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530.
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991, 7, 45–49.
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965.
- Gregg, C.; Zhang, J.; Weissbourd, B.; Luo, S.; Schroth, G.P.; Haig, D.; Dulac, C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 643–648.
- Higgs, M.J.; Hill, M.J.; John, R.M.; Isles, A.R. Systematic investigation of imprinted gene expression and enrichment in the mouse brain explored at single-cell resolution. BMC Genom. 2022, 23, 754.
- Broad, K.D.; Keverne, E.B. Placental protection of the fetal brain during short-term food deprivation. Proc. Natl. Acad. Sci. USA 2011, 108, 15237–15241.
- Davies, W.; Lynn, P.M.; Relkovic, D.; Wilkinson, L.S. Imprinted genes and neuroendocrine function. Front. Neuroendocr. 2008, 29, 413–427.
- Xie, Y.; Dorsky, R.I. Development of the hypothalamus: Conservation, modification and innovation. Development 2017, 144, 1588–1599.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
31 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No