Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanni Cimmino | -- | 4855 | 2023-08-29 13:37:39 | | | |
| 2 | Rita Xu | Meta information modification | 4855 | 2023-08-30 03:52:34 | | | | |
| 3 | Rita Xu | Meta information modification | 4855 | 2023-08-30 03:57:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cimmino, G.; Natale, F.; Alfieri, R.; Cante, L.; Covino, S.; Franzese, R.; Limatola, M.; Marotta, L.; Molinari, R.; Mollo, N.; et al. Non-Conventional Risk Factors in Cardiovascular Disease Prevention. Encyclopedia. Available online: https://encyclopedia.pub/entry/48597 (accessed on 25 July 2026).
Cimmino G, Natale F, Alfieri R, Cante L, Covino S, Franzese R, et al. Non-Conventional Risk Factors in Cardiovascular Disease Prevention. Encyclopedia. Available at: https://encyclopedia.pub/entry/48597. Accessed July 25, 2026.
Cimmino, Giovanni, Francesco Natale, Roberta Alfieri, Luigi Cante, Simona Covino, Rosa Franzese, Mirella Limatola, Luigi Marotta, Riccardo Molinari, Noemi Mollo, et al. "Non-Conventional Risk Factors in Cardiovascular Disease Prevention" Encyclopedia, https://encyclopedia.pub/entry/48597 (accessed July 25, 2026).
Cimmino, G., Natale, F., Alfieri, R., Cante, L., Covino, S., Franzese, R., Limatola, M., Marotta, L., Molinari, R., Mollo, N., Loffredo, F.S., & Golino, P. (2023, August 29). Non-Conventional Risk Factors in Cardiovascular Disease Prevention. In Encyclopedia. https://encyclopedia.pub/entry/48597
Cimmino, Giovanni, et al. "Non-Conventional Risk Factors in Cardiovascular Disease Prevention." Encyclopedia. Web. 29 August, 2023.
Copy Citation
Cardiovascular diseases (CVDs), such as arterial hypertension, myocardial infarction, stroke, heart failure, atrial fibrillation, etc., still represent the main cause of morbidity and mortality worldwide. They significantly modify the patients’ quality of life with a tremendous economic impact. It is well established that cardiovascular risk factors increase the probability of fatal and non-fatal cardiac events. These risk factors are classified into modifiable (smoking, arterial hypertension, hypercholesterolemia, low HDL cholesterol, diabetes, excessive alcohol consumption, high-fat and high-calorie diet, reduced physical activity) and non-modifiable (sex, age, family history, of previous cardiovascular disease).
cardiovascular diseases
conventional risk factors
cardiovascular prevention
1. Introduction
Despite tremendous advancements in prevention and treatment, CVDs are still the leading causes of mortality and the major contributors to disability in industrialized countries, with a huge impact on social and economic systems. Since the first observations from the Framingham Heart Study started in 1948 [1], several other epidemiological studies have confirmed the impact of the so-called conventional CV risk factors, such as age, blood pressure, glucose blood levels, lipid profile, and smoking status, as major determinants of CV disease development and clinical outcome [2]. Based on all these data, the current guidelines on cardiovascular prevention using the SCORE algorithm define the risk of fatal and non-fatal events in a 10-year period [3]. The achievement of targets for all the modifiable risk factors is the primum movens in prevention [3]. However, despite the major effort in promoting a healthy lifestyle and keeping the cardiovascular risk factors at target, in 2019, an estimated 17.9 million people died from CVDs, representing 32% of all global deaths. Of these deaths, 85% were related to heart attack and stroke [4][5][6][7]. Thus, the optimistic expectation of cardiologists to reduce the CVD burden because of improved prevention strategies and treatment of the modifiable risk factors has been largely unmet. Several aspects should be taken into account to explain the reasons of such failure. In December 2022, the American College of Cardiology (ACC) announced the publication of “The Global Burden of Cardiovascular Diseases and Risk: A Compass for Future Health”. In this document, 18 specific CV conditions and 15 risk factors across 21 global regions were analyzed to provide an up-to-date overview of the global burden of CVD [8]. This document includes data from 204 countries, analyzing the major global modifiable CVD risk factors, how they contribute to disease burden, and recent strategies for prevention [8]. Based on this analysis, hypertension, hypercholesterolemia, dietary lifestyle, and air pollution were the leading causes of CVD worldwide. A total of 15 leading risks for CV diseases were included and divided in three categories: environmental (air pollution, household air pollution, low and high temperature); metabolic (systolic blood pressure, low-density lipoprotein cholesterol, body mass index, fasting plasma glucose, kidney dysfunction); and behavioral (dietary, smoking, alcohol use, physical activity). This report has also evaluated the disability-adjusted life years (DALYs), looking at the years of life lost because of premature mortality, and years lived with disability [8]. As a main result of this analysis, ischemic heart disease remains the major cause of CV death, with up to 9.44 million deaths in 2021 and 185 million DALYs. Hypertension remains the modifiable risk factor mainly associated with premature CV deaths, with up to 10.8 million CV deaths and 11.3 million deaths overall in 2021 [8]. A dietary lifestyle evaluation has considered under-consumed food, such as vegetables, fruits, fiber, vegetables, and over-consumed food, such as meats, sodium, and sugar-sweetened beverages. This analysis reveals an association of 6.58 million CV deaths and 8 million deaths overall in 2021 [8]. However, the conventional risk factors evaluated in this latest document may explain only part of the cardiovascular disease burden. In the last few years, several epidemiological and experimental studies have linked the development of CVDs to novel and emerging risk factors [9], such as homocysteine and vitamin D levels, gut microbiota, sleep apnea, sleep duration, uric acid plasma concentration over the air pollution, and climate change, as already stated by the ACC document [8].
2. Metabolic Risk Factors
2.1. Homocysteine: The Never-Ending Debate in Cardiovascular Prevention
Homocysteine is a sulphur amino acid that originates from the metabolism of methionine. Methionine, an essential food-derived amino acid, plays a vital role in cellular processes through the donation of methyl groups [10]. The first metabolite originating from methyl transfer is S-adenosyl methionine, which is subsequently converted to S-adenosyl homocysteine, the immediate precursor of homocysteine. The latter can be ‘recycled’ by taking the methylation route, resulting in the regeneration of methionine, or alternatively, it can be eliminated renally via the transulfuration route, leading to the formation of cysteine. Both processes are mediated by enzymes whose cofactors are vitamin B12, folic acid, and vitamin B6 [11][12]. Under physiological conditions, there is a balance between homocysteine formation and elimination [12]. If homocysteine can accumulate in the body, the biochemical transformation process fails, leading to a serum level increase [12]. Serum homocysteine values between 5 and 15 micromol/L are considered normal while mild hyperhomocysteinemia is defined as values between 15 and 30 micromol/L; moderate, between 30 and 100 micromol/L; and severe, if greater than 100 micromol/L [13]. In the healthy population, blood levels of homocysteine do not appear to be significantly influenced by dietary intake [14]. Hyperhomocysteinemia has many causes, with genetic profiles playing a dominant role: several genetic polymorphisms have been recognized [15] as responsible for the deficiency of enzymes involved in homocysteine metabolism [16]. The most frequent polymorphisms involve the gene coding for methylenetetrahydrofolate reductase and the one coding for cystathionine beta synthase [17]. Other causes include vitamin B12, B6 and folic acid deficiency [18]; advanced age; male sex; menopause; lifestyle habits, such as alcohol abuse and smoking [19]; and certain diseases, including cancers [15], chronic kidney disease [20], hypothyroidism [21], and inflammatory bowel disease [22]. Mention should be made of drugs that may interfere with the metabolism of homocysteine or its enzymatic cofactors: these include methotrexate, carbamazepine, nitrates, fibrates, and metformin [23].
Over the past few decades, the correlation of homocysteine with the incidence of cardio- and cerebrovascular events as well as its potential role in the pathogenesis of atherosclerosis have been the subject of countless debates [24][25][26]. The first correlation between serum homocysteine levels and the incidence of coronary artery disease is dated 1956 [27]. Numerous clinical studies and meta-analyses have subsequently supported this theory, reporting a 20% increase in the risk of new coronary events for every 5 micromol/L increase above normal serum homocysteine levels [28] and an increased risk of fatal and non-fatal coronary [29][30][31] and cerebrovascular events [30][32]. Further analyses corroborate these data, showing a 25% reduction in homocysteine levels (approximately 3 micromol/L) correlates with a lower risk of cardiac ischemic events and stroke [32].
The relationship between hyperhomocysteinemia and mortality for coronary artery diseases or cardiovascular causes or all causes has been evaluated in a meta-analysis of 20 prospective studies reporting that elevated homocysteine levels were an independent predictor of cardiovascular events, mortality from cardiovascular causes, and mortality from all causes [33].
Other studies have correlated hyperhomocysteinemia with an increased risk for and recurrence of venous thromboembolic events [34][35][36], peripheral artery diseases [37], and congestive heart failure [38].
Based on this evidence, hyperhomocysteinemia has been proposed as an independent cardiovascular risk factor [38][39].
Several cellular mechanisms have been proposed to explain how hyperhomocysteinemia is implicated in the etiology of cardio- and cerebrovascular events. Endothelial dysfunction, increased arterial stiffness, and a prothrombotic state are common in patients with hyperhomosysteinemia [40]. The main pathways associated with this endothelial impairment are: a) increased oxidative stress [41]; b) a reduction in the expression of the endothelial isoform of nitric oxide synthetase (eNOS) and increase in the cellular expression of caveolin-1 that is an inhibitor of eNOS, thus leading to a reduced release of nitric oxide [42]; and c) the upregulation of cell adhesion molecules, resulting in an increased chemotaxis of monocytes on the endothelium and increased endothelial expression of IL-8, which favor inflammatory processes [43].
Hyperhomocysteinemia is also associated to collagen synthesis [44] and vessel smooth muscle cell proliferation [45], through activation of cyclin A, protein kinase C, and the proto-oncogenes c-myc and c-fos [45][46] as well as increased production of phospholipids [46] and increased expression of platelet growth factor [47]. This smooth muscle cells proliferation as well as increased collagen deposition and alterations in elastic tissue composition [48] is responsible for increased arterial wall stiffness [49][50][51]. This phenomenon is facilitated by the inactivation of eNOS and the reduced production of nitric oxide [52]. A schematic view of homocysteine pathways involved in CVD is provided in Figure 1.
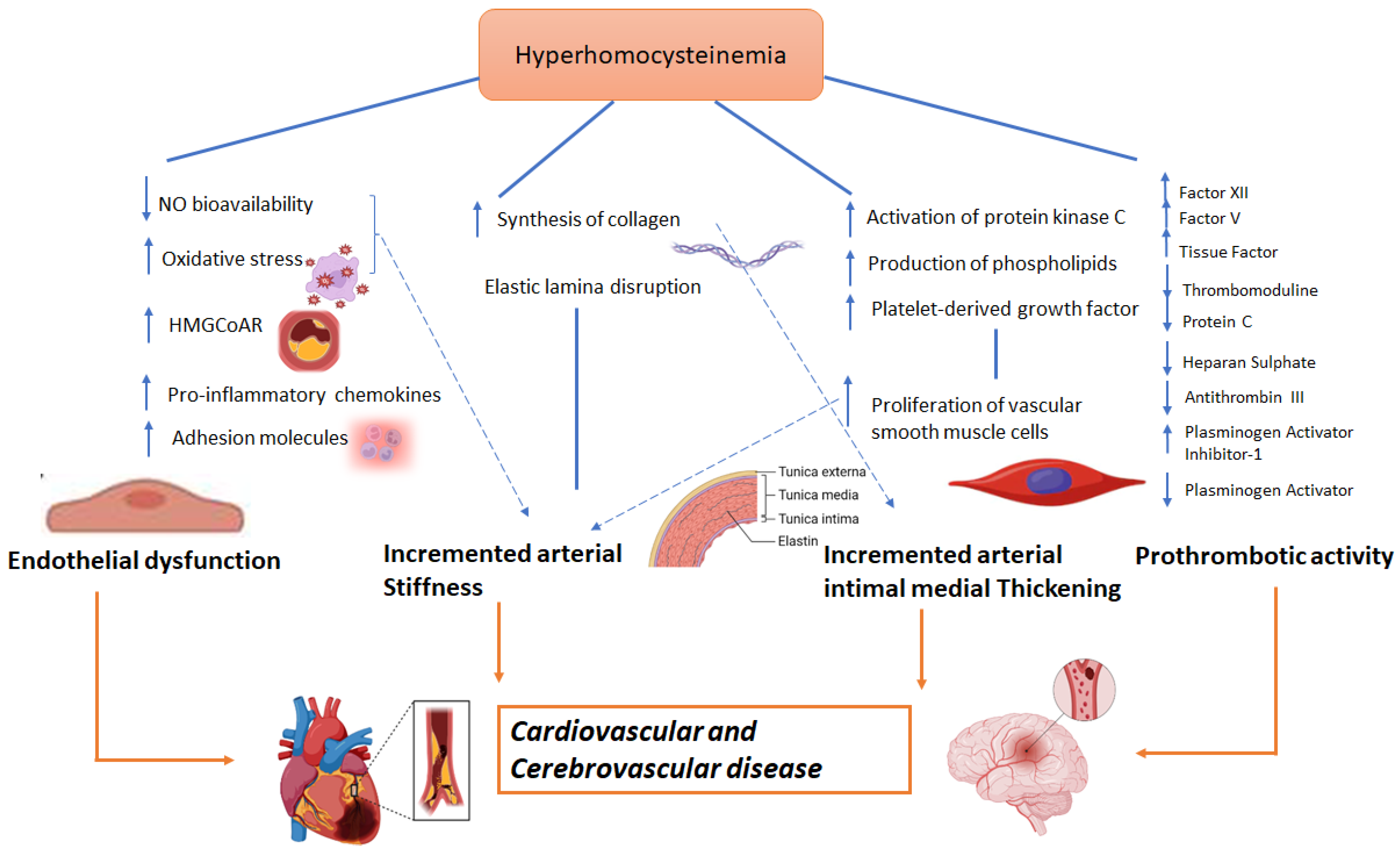
Figure 1. Possible role of homocysteine in CVD.
Moreover, several studies have also linked hyperhomocysteinemia to increased prothrombotic state [53]. This effect has been mainly related to: (a) factor XII and factor V activation [54]; (b) tissue factor expression [55]; (c) thrombomodulin inhibition [56] that results in a reduction of protein C activation [57]; (d) a reduction in the anticoagulant effect of antithrombin III, thus altering the binding capacity of endothelial heparan sulphate with the latter [58]; and (e) the reduction of plasminogen activator function and increased expression of its inhibitor [59].
In light of these basic findings, several clinical studies have investigated whether the treatment of hyperhomocysteinemia might result in cardiovascular benefits in terms of cardio- and cerebrovascular event reduction with conflicting results.
2.2. Uric Acid: Still a Controversial Cardiovascular Risk Factor?
Uric acid (UA) is the final product of purine metabolism. The increase in its blood levels may depend either on an increased production or on a reduced elimination [60]. If hyperuricemia develops, urate crystals accumulation may occurs in the joints leading to the clinical manifestations of gout, subsequently also affecting the renal parenchyma and the excretory tracts with the picture of gouty nephropathy and nephro/urolithiasis [61]. Beyond this known effect, several other clinical studies have also investigated the relationship between high blood levels of UA and the development of CVDs [62] and, as for homocysteine, with conflicting results. The Framingham Heart Study did not indicate hyperuricemia as an independent risk factor for coronary artery disease, cardiovascular death, and death from all causes [63][64]. Some epidemiological studies have described a J- or U-shaped relationship between UA levels and cardiovascular risk, meaning that patients with either very low or very high UA values have an increased cardiovascular risk [65]. More recently, clinical studies seem to support the role of hyperuricemia in atherosclerosis, systemic arterial hypertension, atrial fibrillation, and chronic kidney disease as the pathophysiological processes promoted by UA, such as oxidative stress and inflammation that are the basis of endothelial dysfunction, which may contribute to atherothrombotic events. An increase in the activity of the enzyme xanthine oxidase, which regulates the synthesis of UA and which uses molecular oxygen as an electron acceptor for its function, determines the formation of reactive oxygen species (ROS) [66]. ROS are responsible for the lipid oxidation and the reduction of the nitric oxide concentration, which causes the loss of the physiological vasodilating effect of the endothelium and determines a prothrombotic phenotype. UA also favors an increase in the deposition of low-density lipoproteins at the endothelial level and their uptake by macrophages, which are transformed into foam cells, thus starting the process of atherosclerosis [67]. More recently, it has been highlighted how endothelial cells (ECs) may acquire a prothrombotic phenotype by expressing functional tissue factor (TF) once exposed to increasing doses of UA that can be reversed by the preincubation with an uricosuric agent [68]. Moreover, the endothelial dysfunction induced by hyperuricemia also favors the expression on the cell surface of the adhesion molecules (CAMs) involved in the initiation of the atherosclerosis process. This mechanism appears to be regulated by a modulation of the NF-kappaB pathway, leading to the upregulation of TF on cell surface and downregulation of its natural inhibitor, the Tissue Factor Pathway inhibitor (TFPI) [69]. Furthermore, the inflammasome [70] seems also to be involved with an increase in caspase-1 function, which would promote a particular type of endothelial cell apoptosis, known as pyroptosis, and the release of TNF-alpha [71]. A summary of the possible mechanisms by which UA is involved in CVD is provided in Figure 2.
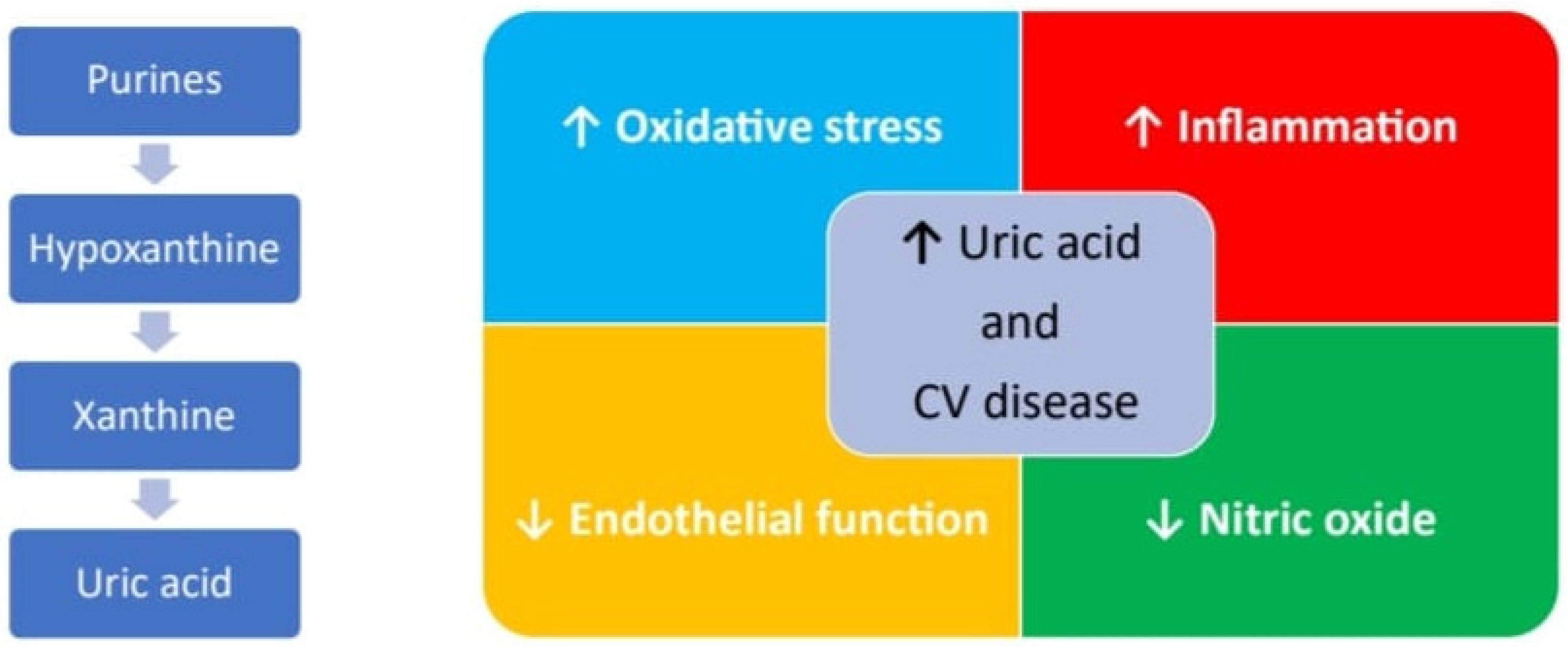
Figure 2. Major pathways UA related involved in pathogenesis of CVD.
These basic findings have been corroborated by a more recent clinical evaluation on patients with acute coronary syndrome (ACS) [72] by reporting that higher UA levels are associated with higher C-reactive protein (CRP) and troponin values. Additionally, ACS patients with high UA levels showed an angiographic picture of multivessel coronary artery disease and complex atherosclerosis according to the Ellis classification [72]. As regards the relationship between hyperuricemia and systemic arterial hypertension, several studies have shown an increase in blood pressure in patients with increased uric acid. A meta-analysis that studied 55,607 patients showed that for each 1 mg/dL increase in uric acid, the incidence of arterial hypertension increases by approximately 13% [73]. At the basis of this relationship, there would be the lower release of nitric acid and the activation of the renin–angiotensin–aldosterone system promoted by uric acid, which determine vasoconstriction and consequent increase in blood pressure. A relationship between hyperuricemia and increased onset of atrial fibrillation (AF) has been highlighted by the ARIC study, which shows a 1.16-fold increase in the risk of AF in subjects, mostly female and of African origin, with high UA values [74]. Atrial remodeling induced by the inflammatory effects and oxidative stress related to UA seems to be the underlying mechanism [75]. In light of the relationship between hyperuricemia and increased cardiovascular risk, the current therapeutic options mainly are represented by allopurinol and febuxostat, which inhibit the enzyme xanthine oxidase, and therefore, the UA production could have a role in reducing the incidence of cardiovascular events.
2.3. Vitamin D: Light and Shadow in Cardiovascular Prevention
Vitamin D, commonly known as the “sunshine vitamin”, is an essential nutrient that plays a critical role in the absorption and regulation of calcium and phosphorus, essential minerals necessary for strong bones, teeth, and overall skeletal health [76]. Unlike other vitamins, the human body can produce vitamin D through exposure to sunlight [77]. The precursor form of vitamin D, indeed known as 7-dehydrocholesterol, is naturally present in the skin [78]. Upon exposure to UVB radiation emitted by sunlight, a photochemical reaction takes place, leading to the transformation of 7-dehydrocholesterol into pre-vitamin D3 [78]. Subsequently, through heat-induced isomerization, pre-vitamin D3 is converted into cholecalciferol, also known as vitamin D3. Another form of vitamin D, the Vitamin D2, also known as ergocalciferol, is primarily derived from plant-based sources and is commonly utilized in fortified food products and some dietary supplements. Vitamin D2 and D3 are fully activated through two consecutive hydroxylation reactions catalyzed by specific P450 isoenzymes. The First hydroxylation, which occurs on the carbon in position 25, takes place in the liver by vitamin D 25-hydroxylase (CYP2R1) to form the pro-hormone 25-hydroxyvitamin D. Due its solubility and BPD binding properties, the level of this metabolite better reflects the body’s vitamin D status. The second hydroxylation occurs on the carbon in position 1 by 25-hydroxyvitamin D-1alpha-hydroxylase renal (CYP27B1) and is responsible for the synthesis of the biologically active metabolite, 1,25-dihydroxyvitamin D [78].
Beyond its well-known role in bone health, vitamin D has garnered increasing attention in relation to cardiovascular health. Numerous observational studies have investigated the link between vitamin D levels and CVDs. Although the results show some degree of variability, they consistently highlight an inverse association between vitamin D status and the risk of developing CVD [79][80][81]. The inverse correlation between vitamin D status and CVD seems to be particularly strong in older adults [82][83]. Meta-analyses of epidemiological studies support the inverse correlation between vitamin D levels and CVD [82][84]. The correlation between vitamin D levels and arterial hypertension holds significant importance. Blood pressure tends to exhibit geographical and racial disparities, whereby the risk of hypertension tends to rise from south to north in the Northern hemisphere. A suggested explanation for this latitude-based correlation is that sunlight exposure may offer protection, potentially due to the influence of ultraviolet B (UVB) radiation or vitamin D [85]. This association appears to be supported by animal studies. Mice that lack the vitamin D receptor (VDR) or have a genetic deficiency in the 1-alpha-hydroxylase gene, which is responsible for vitamin D activation, have been shown to develop high renin hypertension and cardiac hypertrophy [86][87]. In vitro studies highlight a favorable cardioprotective effect of 1,25-dihydroxyvitamin D. It has been reported that the pretreatment of ECs with vitamin D reduce the expression and activity of TF and CAMs induced by oxidized lipids [68] or interleukin-6 [88], possibly preserving endothelial function.
All the putative cardiovascular mechanisms associated with vitamin D are provided in Figure 3.
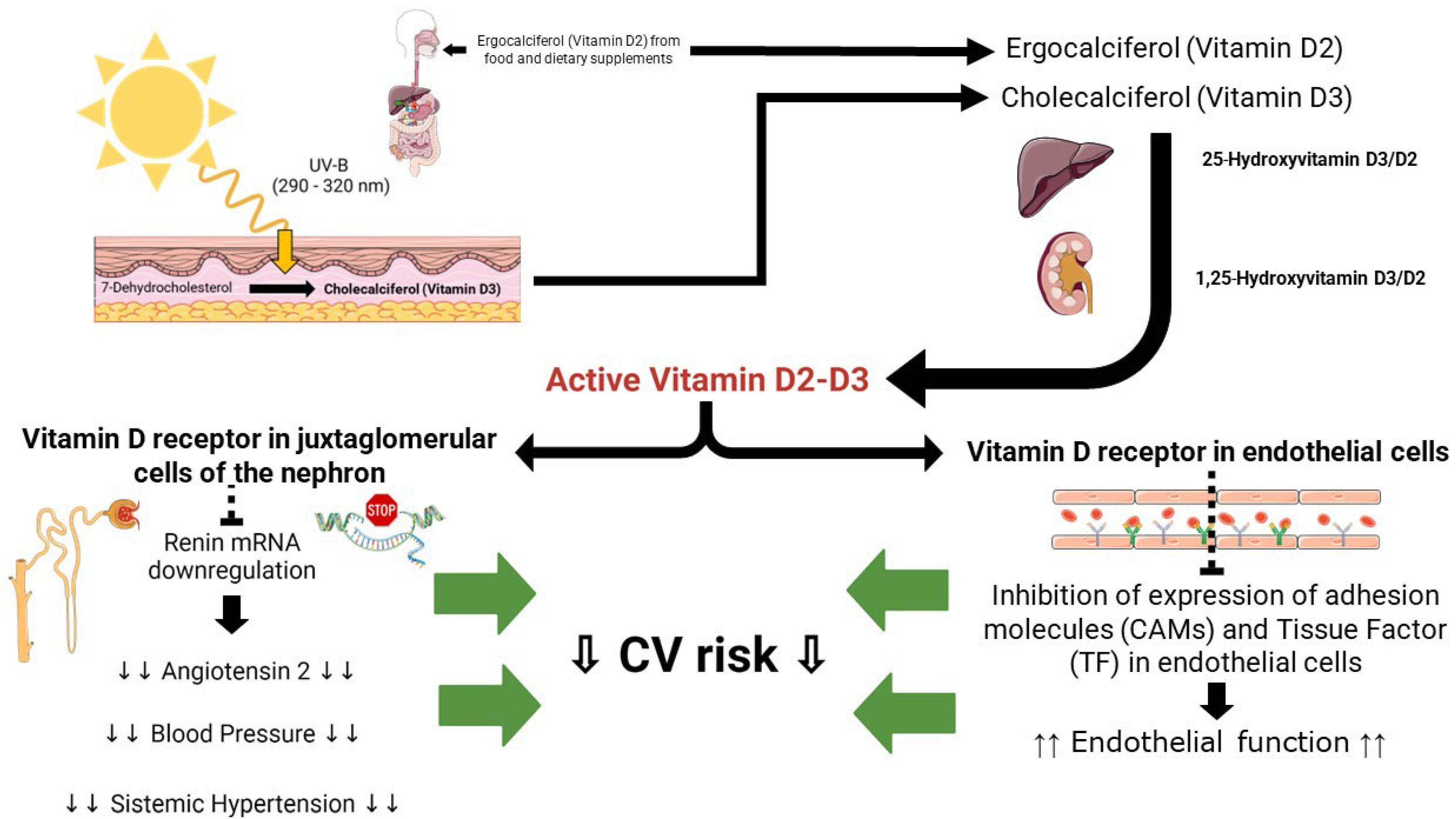
Figure 3. Putative cardiovascular pathways Vitamin D-related: see text for details.
While in vitro studies and epidemiological studies have provided promising insights into the potential cardioprotective effects of vitamin D, the results from randomized controlled trials (RCTs) in this field have been inconclusive to date. The majority of trials conducted so far have primarily focused on investigating the impact of vitamin D supplementation on bone health. In many cases, vitamin D supplementation has been administered alongside calcium supplementation. Meta-analyses of randomized controlled trials (RCTs) have demonstrated non-significant reductions in CVD events with vitamin D supplementation [89][90][91].
3. Non-Metabolic Risk Factors and Surrogates
3.1. Obstructive Sleep Apnea Syndrome: The Diving Board to CVDs
Obstructive sleep apnea (OSA) syndrome is a clinical condition characterized by cyclical episodes of total (apnea) or partial (hypopnea) collapse of the upper airways, occurring during sleep, with the persistence of thoracoabdominal movements. At the end of the events, arousal occurs with transient hypoxemia, autonomic alterations, and sleep fragmentation [92].
Apnea is defined as a reduction in airflow of at least 90% compared to the basal one, lasting at least 10 s while hypopnea is defined as a reduction in airflow of at least 30%, for no less than 10 s, associated with a reduction of at least 3% in oxygen saturation (SaO2) [93].
The severity of OSA is based on the number of events/hour, and it is defined as AHI index (apnea/hypopnea index). Specifically, <5 events/hour define a normal respiratory pattern, 5–14 events/hour a mild apnea, 15–29 events/hour a moderate apnea, and from 30 events/h a severe apnea [93]. The gold standard for the diagnosis of OSA is represented by polysomnography (PSG) [93].
A diagnosis of OSA is made based on nocturnal breathing disorders (snoring, breathing pauses in sleep, restless sleep, awakening choking) and/or daytime sleepiness symptoms associated with an AHI > 5; on the contrary, if the AHI index is greater than 15, OSA can be diagnosed in the absence of symptoms [94].
In general population, OSA prevalence is approximately 34% in men and 17% in women [92][95] while in CVD populations, it ranges from 40% to 60% [96][97].
During sleep, a failure of the neuromuscular reflex that preserves the patency of the airways occurs, resulting in hypoxemia and hypercapnia, determining an increase in the respiratory effort and an awakening of a few seconds, which restores patency of the upper airways, thanks to a series of reflex mechanisms. When sleep resumes, the cycle repeats [98].
OSA represents an independent risk factor for CVDs, such as HTN, AF and other arrhythmias, HF, CAD, stroke, pulmonary hypertension, metabolic syndrome, and diabetes as shown in Figure 4. The involved mechanisms are multiple and probably interconnected.
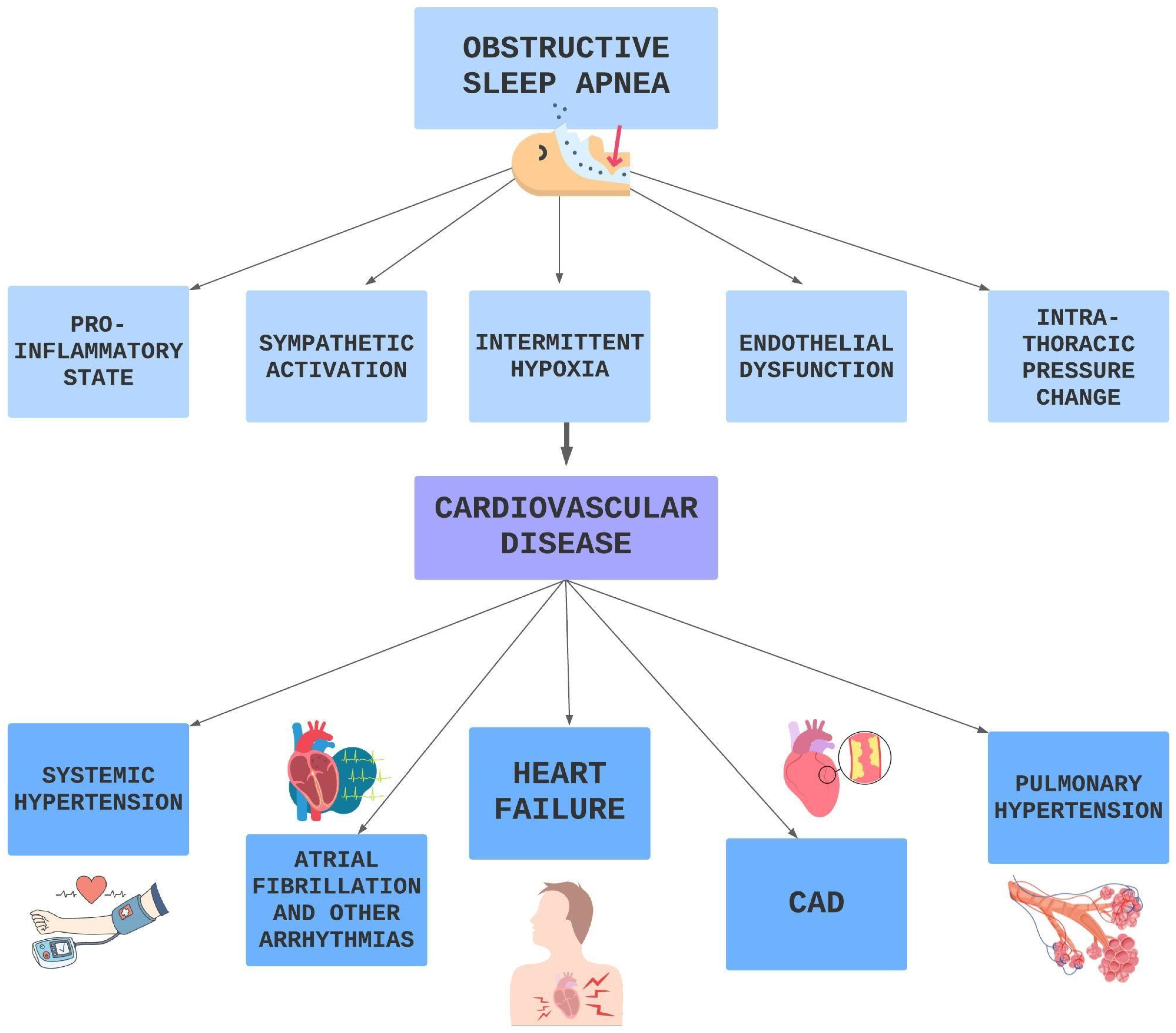
Figure 4. Pathophysiological pathways OSA related leading to CVD.
During the apneic phase, by stimulating peripheral and central chemoreceptors [99], hypoxia and hypercapnia determine the activation of the sympathetic nervous system with consequent peripheral vasoconstriction and an increase in vascular resistance and heart rate [100]. This results in an increase in left ventricular afterload and cardiac work. In addition, there is an overall increase in left ventricular transmural pressure (that is the difference between ventricular systolic pressure and intrathoracic pressure) with increased wall stress [101][102]. The cycle repeats many times every night; therefore, the cardiovascular system is chronically exposed to neuro-hormonal stress, and the hyperactivity of the autonomic nervous system also extends to the daytime hours over time [100][103].
Intermittent hypoxia is also responsible for an increase in oxidative stress [104]: during the hypoxic phase, the cells adapt to an environment with low oxygen content, and with the reoxygenation phase, there is a sudden increase of oxygen with ROS formation, leading to cellular damage in the ischemic tissue [105][106].
Furthermore, a reduction in the levels of circulating NO has also been highlighted during OSA [107], and this could be implicated in endothelial dysfunction [108].
OSA is present in up to 30–50% of HTN patients, and 80% of patients with resistant HTN have OSA [94][109], representing an independent risk factor [92]. In patients with OSA, due to the overactivity of the sympathetic nervous system, the physiological reduction in blood pressure during the night (which configures the “dipper” profile) does not occur [110][111]. Therefore, there seems to be a correlation between sleep apnea and the non-dipper profile of essential HTN [112][113]. Furthermore, several randomized trials and meta-analysis have shown a reduction in blood pressure in patients with sleep apnea treated with CPAP [92][114].
OSA is associated with heart rhythm disturbances and sudden death; pauses and bradycardia are common in patients with OSA [94].
OSA is also an independent risk factor for AF with several pathophysiological mechanisms implicated. In particular, sudden changes in intrathoracic pressure can cause atrial remodeling and atrial fibrosis with consequent electrophysiological alterations [115]. Moreover, the sudden increase in sympathetic activity during apneas can lead to the activation of catecholamine-sensitive atrial on channels, thus determining focal discharges from which AF can be originated [116]. OSA is also associated with an increase in systemic inflammation, which may contribute to the genesis of AF [117].
Sleep apnea also increases the risk of CAD by favoring atherosclerotic process via oxidative stress, endothelial dysfunction, inflammatory state, and autonomic dysfunction. It has been reported that in OSA patients, myocardial infarction occurs more frequently during the night hours [118], and a higher pro-inflammatory profile is present [119] with an effective reduction of the latter if CPAP therapy is used [119]. This study, therefore, suggests that OSA could activate vascular inflammation with non-traditional pathogenetic mechanisms.
OSA is also a risk factor for incident strokes, stroke recurrence [120], and functional and cognitive outcomes [121].
Pulmonary hypertension is closely related to OSA. Hypoxia and hypercapnia induce arteriolar vasoconstriction in the short term and vascular remodeling in the long term that could lead to an irreversible increase in pulmonary vascular resistance and the development of pulmonary hypertension [122].
Sleep apnea, mainly the central form (CSA), is highly prevalent in HF patients as well, ranging from 40% to 60% of symptomatic patients [123].
OSA is also linked to obesity and metabolic syndrome since chronic intermittent hypoxemia and sleep loss is associated to higher plasma leptin levels [124], glucose metabolism impairment, and insulin resistance [125].
At least, there is a reciprocal interaction between obesity and OSA where they both reinforce their progression and their severity in a vicious circle. It is believed that the deposition of fat in the upper airways and the functional alteration of the airways themselves are the mechanisms involved in the pathogenesis of OSA in the obese subjects [126]. On the other hand, daytime sleepiness and decreased physical activity together with hyperleptinemia are the mechanisms probably implicated in weight gain in OSA.
3.2. Air Pollution: Health Breath as Part of Prevention
Air pollution is the contamination of the environment, indoor or outdoor, by a mixture of chemical, physical, or biological agents that change the characteristics of the atmosphere and even at low concentrations cause damage to human health, other living organisms and the environment [127]. According to the Global Burden of Disease (GBD) report, air pollution was responsible for 6.7 million deaths in 2019 alone [127][128]. Globally, nearly 20% of CVD deaths are attributable to air pollution [128]. The main components of this mixture of pollutants are Total Suspended Particulate Matter (PM), gaseous compounds including ozone (O3), nitrogen dioxide (NO2), carbon monoxide (CO), sulfur dioxide (SO2), and volatile organic compounds including benzene [127]. According to the World Health Organization, 99% of the world’s population breathes air that contains annual average levels of air pollutants that exceed guideline recommendations. Particularly high exposures have been documented in cities in Asia, western sub-Saharan Africa, and Latin America [127]. The most consistent evidence on health damage is attributed to PM, i.e., the set of airborne particles, ranging in diameter from 0.1 to 100 mm, capable of remaining in suspension in the air even for long periods [129][130]. Short- and long-term exposure to PM is associated with increased morbidity and mortality, impacting the progression of atherosclerosis [131], ischemic heart disease [132][133][134], stroke [135], and lung disease as well as the course of pregnancy and the health of newborns [136]. PM10 (particles between 2.5 and 10 mm in diameter) and, largely, PM2.5 (diameter < 2.5 mm), are the most linked to CVD and affecting global public health [137][138]. Lung inflammation and oxidative stress pathway is the primary response to air pollution exposure [139], contributing to the development of a systemic pro-inflammatory state and activation of secondary effector pathways that result in endothelial dysfunction, increased atherosclerotic plaque vulnerability, and the activation of a prothrombotic and proarrhythmic state [132][140][141]. Experimental animal models seem indeed to support this hypothesis [142]. Moreover, human exposure to pollutant nanoparticles causes their translocation into the systemic circulation through the alveolus-capillary membrane, interacting with the endothelium, accumulating at sites of vascular inflammation, thus favoring atherosclerotic process [143][144][145], with effects similar to those observed in the lungs [146] and thrombotic complications [147]. A relevant change in platelet function toward increased prothrombotic tendency has been confirmed in diabetic patients after recent (within two hours) exposure to PM [148]. In addition to these mechanisms, short-term PM2.5 exposure in animal models is associated with sympathetic nervous system activation and hypertension, probably mediated by neuroinflammation [149][150]. In a meta-analysis of 33 studies, short-term exposure to PM2.5 was associated with a significant decrease in heart rate variability (HRV) [151]. Decreased HRV is an index of autonomic system dysfunction and predicts an increased risk of cardiovascular morbidity and mortality in patients with heart disease [152]. Increased blood pressure and decreased HRV suggest an autonomic imbalance in favor of sympathetic tone and could further explain the rapid cardiovascular responses associated with air pollution, such as the initiation of fatal tachyarrhythmias and increased myocardial infarctions [132], as confirmed by the available literature [153]. High short-term exposure to PM2.5 is associated with an increased risk of acute coronary event, acutely destabilizing and rupturing atherosclerotic plaque, in patients with clinically significant pre-existing CAD but not in those with uninjured coronary arteries [154]. Moreover, short-term exposure to elevated levels of PM2.5 and PM10 is also associated with increased daily hospitalizations for STEMI and increased incidence of STEMI-related ventricular arrhythmias and cardiac death [155]. The effect of long-term exposure to major air pollutants was assessed by the ESCAPE study that have evaluated the incidence of acute coronary events in 11 European cohorts. At a mean follow-up of 11.5 years, exposure to annual mean levels of PM2.5 > 5 μg/m3 and PM10> 10 μg/m3 was associated with a 13% and 12% increase in the risk of nonfatal acute coronary events, respectively, with no evidence of heterogeneity between cohorts [156]. Other observational studies and meta-analyses have reported a positive correlation between long-term exposure to air pollution and the development and progression of subclinical atherosclerosis and calcium accumulation [157] as well as increased carotid intima-media thickness [158]. Based on the published data, no more doubts should exist on the role of air pollutants in CVD development. A schematic view of the relationship between air pollution and CVD is provided in Figure 5.
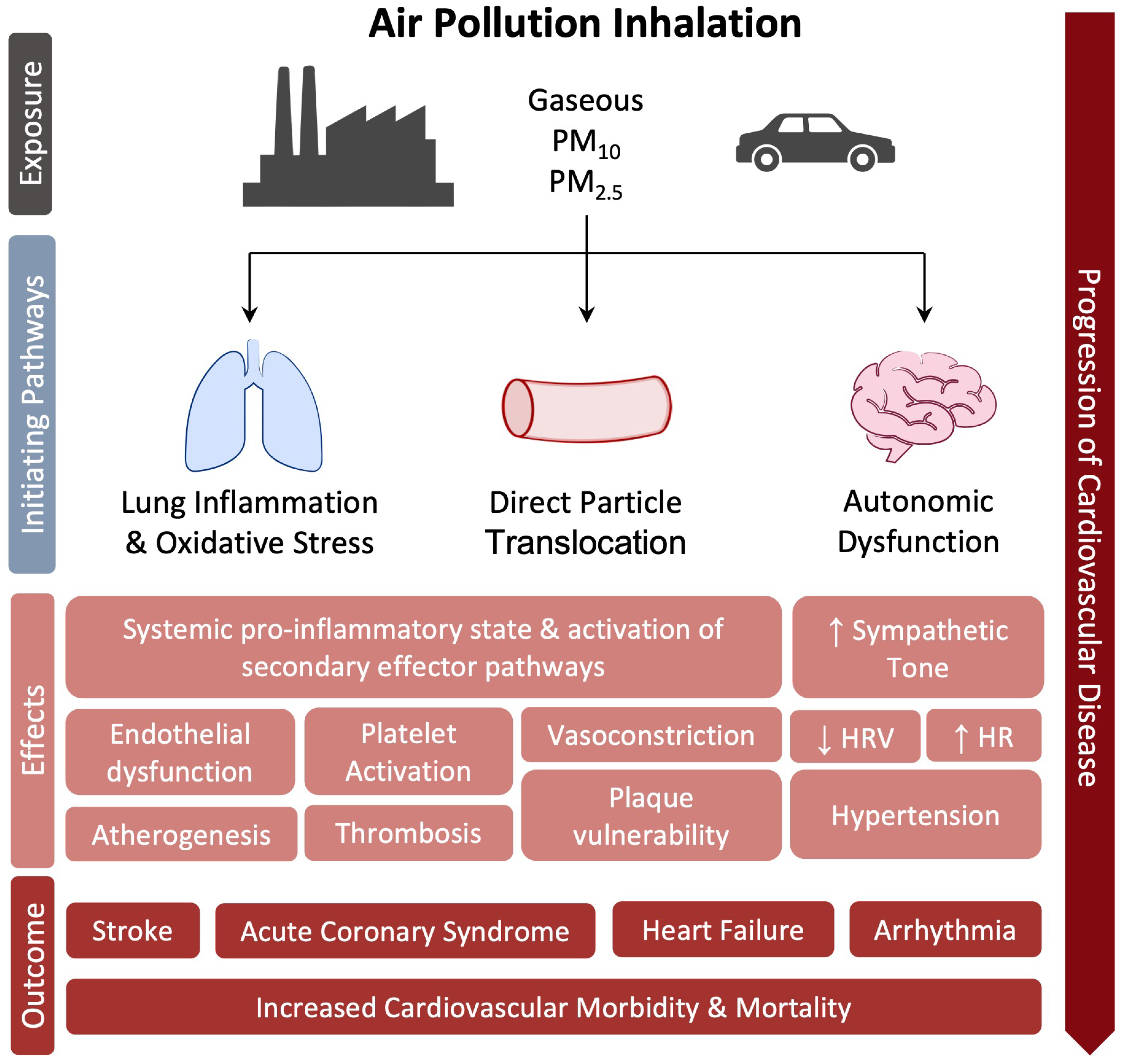
Figure 5. Molecular mechanisms linked air pollution to CVD.
3.3. Climate Change: The Impact of Temperature
Temperature and its extreme variation is now recognized as a cardiovascular risk factor [159][160][161]. A very recent analysis evaluating 32,000 cardiovascular deaths in 27 countries on 5 continents over 40 years support the role of extremely hot or cold temperatures in determining heart disease deaths [161]. Mortality and morbidity induced by climate change are not exclusively due to hypothermia or hyperthermia, but also to indirect causes, such as respiratory diseases and CVDs, which can be undetected when the human body tries to adapt to climate changes [162]. A relationship between mortality from CVD and temperature exists with a U-, V-, or J shaped curve [163][164][165]. While the correlation between temperature and CVD has been established, the role of diurnal temperature range (DTR), defined as the difference between the maximum and minimum temperatures recorded in one day, in determining CV events needs to be better evaluated. Extreme cold weather conditions associated to climate change contributes to an increase in temperature variability that might increase clinical cardiovascular events [160]. It is known that exposure to cold activates both the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS), which interact with each other, leading to HTN and myocardial damage [166]. Skin blood flow (SBF) is reduced in response to cold due to vasoconstriction and increased urine output, thus inducing dehydration, hemoconcentration, and hyperviscosity [167]. Furthermore, eNOS and adiponectin inhibition contributes to endothelial dysfunction and lipid deposition, thus favoring atherosclerosis and plaque instability. Cold exposure also triggers mitochondrial dysfunction with myocardial damage, cardiac hypertrophy, and cardiac dysfunction. The increase in cardiac work and peripheral resistance contributes to an increase in oxygen consumption and a reduction in the ischemic threshold [166], which is clinically relevant, especially when the coronary circulation is already compromised.
On the contrary, exposure to heat leads to increased blood flow and sweating with loss of fluids and dehydration. The resulting hemoconcentration and hyperviscosity may cause thromboembolism, leading to increased risk of ischemic stroke [168]. In the presence of heat stroke, the increase in core temperature redistributes the flow on the skin to facilitate heat loss. Intestinal blood flow is reduced, and this could cause increased permeability of the intestinal epithelium, allowing bacteria, their toxic cell wall component LPS, or HMBG1 to move from the intestinal lumen into the circulation. TLR4 recognizes these molecules, stimulating innate and adaptive immune responses and causing systemic inflammatory response syndrome (SIRS). Along with this, hyperthermia induces the occlusion of arterioles and capillaries (microcirculatory thrombosis) or excessive bleeding (consumptive coagulation), leading to multiorgan dysfunction. The putative mechanisms linking climate changes and CVD is provided in Figure 6.
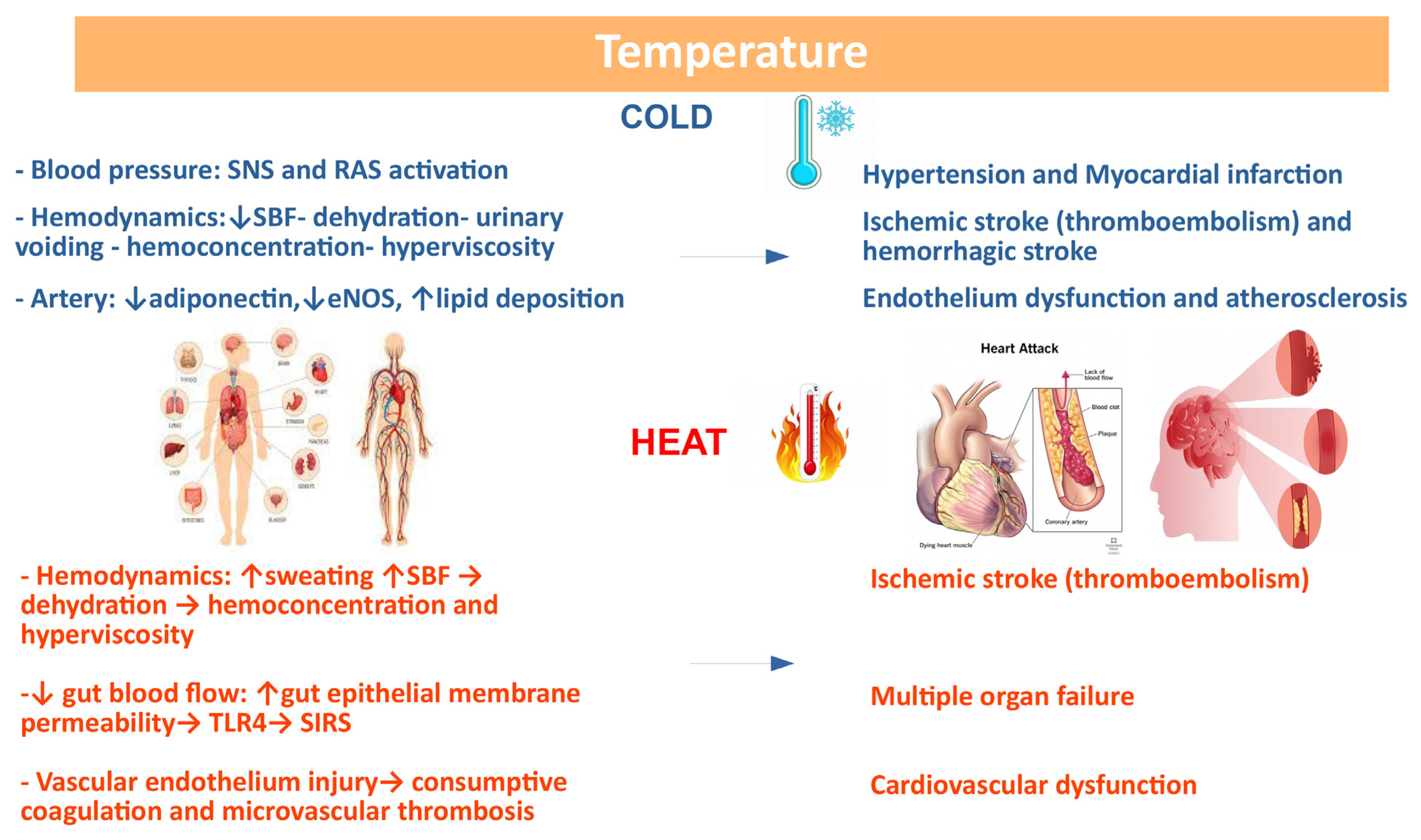
Figure 6. Correlation between climate changes and CVD: possible basic mechanisms. Several variables affect the response to temperature changes.
References
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2014, 383, 999–1008.
- Dzau, V.J.; Antman, E.M.; Black, H.R.; Hayes, D.L.; Manson, J.E.; Plutzky, J.; Popma, J.J.; Stevenson, W. The Cardiovascular Disease Continuum Validated: Clinical Evidence of Improved Patient Outcomes. Circulation 2006, 114, 2850–2870.
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337.
- Townsend, N.; Kazakiewicz, D.; Wright, F.L.; Timmis, A.; Huculeci, R.; Torbica, A.; Gale, C.P.; Achenbach, S.; Weidinger, F.; Vardas, P. Epidemiology of cardiovascular disease in Europe. Nat. Rev. Cardiol. 2022, 19, 133–143.
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596.
- Hackam, D.G. The Changing Epidemiology of Cardiovascular Disease: Two Steps Forward, One Step Back. Can. J. Cardiol. 2020, 36, 995–996.
- Noale, M.; Limongi, F.; Maggi, S. Epidemiology of Cardiovascular Diseases in the Elderly. Frailty Cardiovasc. Dis. Res. Elder. Popul. 2020, 1216, 29–38.
- Lindstrom, M.; DeCleene, N.; Dorsey, H.; Fuster, V.; Johnson, C.O.; LeGrand, K.E.; Mensah, G.A.; Razo, C.; Stark, B.; Varieur Turco, J.; et al. Global Burden of Cardiovascular Diseases and Risks Collaboration, 1990–2021. J. Am. Coll. Cardiol. 2022, 80, 2372–2425.
- Lacey, B.; Herrington, W.G.; Preiss, D.; Lewington, S.; Armitage, J. The Role of Emerging Risk Factors in Cardiovascular Outcomes. Curr. Atheroscler. Rep. 2017, 19, 28.
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell Biol. 2000, 32, 385–389.
- Tchantchou, F. Homocysteine metabolism and various consequences of folate deficiency. J. Alzheimer’s Dis. JAD 2006, 9, 421–427.
- Finkelstein, J.D. The metabolism of homocysteine: Pathways and regulation. Eur. J. Pediatr. 1998, 157 (Suppl. S2), S40–S44.
- Kang, S.S.; Wong, P.W.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298.
- Ubbink, J.B.; Vermaak, W.J.; van der Merwe, A.; Becker, P.J. The effect of blood sample aging and food consumption on plasma total homocysteine levels. Clin. Chim. Act Int. J. Clin. Chem. 1992, 207, 119–128.
- Singal, R.; Ferdinand, L.; Das, P.M.; Reis, I.M.; Schlesselman, J.J. Polymorphisms in the methylenetetrahydrofolate reductase gene and prostate cancer risk. Int. J. Oncol. 2004, 25, 1465–1471.
- Sharma, P.; Senthilkumar, R.D.; Brahmachari, V.; Sundaramoorthy, E.; Mahajan, A.; Sharma, A.; Sengupta, S. Mining literature for a comprehensive pathway analysis: A case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis. 2006, 5, 1.
- Summers, C.M.; Hammons, A.L.; Mitchell, L.E.; Woodside, J.V.; Yarnell, J.W.; Young, I.S.; Evans, A.; Whitehead, A.S. Influence of the cystathionine beta-synthase 844ins68 and methylenetetrahydrofolate reductase 677C>T polymorphisms on folate and homocysteine concentrations. Eur. J. Hum. Genet. EJHG 2008, 16, 1010–1013.
- Siri, P.W.; Verhoef, P.; Kok, F.J. Vitamins B6, B12, and folate: Association with plasma total homocysteine and risk of coronary atherosclerosis. J. Am. Coll. Nutr. 1998, 17, 435–441.
- Refsum, H.; Nurk, E.; Smith, A.D.; Ueland, P.M.; Gjesdal, C.G.; Bjelland, I.; Tverdal, A.; Tell, G.S.; Nygard, O.; Vollset, S.E. The Hordaland Homocysteine Study: A community-based study of homocysteine, its determinants, and associations with disease. J. Nutr. 2006, 136, 1731S–1740S.
- Bostom, A.G.; Lathrop, L. Hyperhomocysteinemia in end-stage renal disease: Prevalence, etiology, and potential relationship to arteriosclerotic outcomes. Kidney Int. 1997, 52, 10–20.
- Sengul, E.; Cetinarslan, B.; Tarkun, I.; Canturk, Z.; Turemen, E. Homocysteine concentrations in subclinical hypothyroidism. Endocr. Res. 2004, 30, 351–359.
- Papa, A.; De Stefano, V.; Danese, S.; Chiusolo, P.; Persichilli, S.; Casorelli, I.; Zappacosta, B.; Giardina, B.; Gasbarrini, A.; Leone, G.; et al. Hyperhomocysteinemia and prevalence of polymorphisms of homocysteine metabolism-related enzymes in patients with inflammatory bowel disease. Am. J. Gastroenterol. 2001, 96, 2677–2682.
- Desouza, C.; Keebler, M.; McNamara, D.B.; Fonseca, V. Drugs affecting homocysteine metabolism: Impact on cardiovascular risk. Drugs 2002, 62, 605–616.
- Cybulska, B.; Kłosiewicz-Latoszek, L. Homocysteine—Is it still an important risk factor for cardiovascular disease? Kardiol. Pol. 2015, 73, 1092–1096.
- Tripathi, P. Homocysteine- The Hidden Factor and Cardiovascular Disease: Cause or Effect? Biochem. Anal. Biochem. 2015, 4, 1000237.
- Brattstrom, L.; Wilcken, D.E. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323.
- Wilcken, D.E.; Wilcken, B. The pathogenesis of coronary artery disease. A possible role for methionine metabolism. J. Clin. Investig. 1976, 57, 1079–1082.
- Humphrey, L.L.; Fu, R.; Rogers, K.; Freeman, M.; Helfand, M. Homocysteine level and coronary heart disease incidence: A systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 1203–1212.
- Drewes, Y.M.; Poortvliet, R.K.; Blom, J.W.; de Ruijter, W.; Westendorp, R.G.; Stott, D.J.; Blom, H.J.; Ford, I.; Sattar, N.; Wouter Jukema, J.; et al. Homocysteine levels and treatment effect in the PROspective Study of Pravastatin in the Elderly at Risk. J. Am. Geriatr. Soc. 2014, 62, 213–221.
- Boushey, C.J.; Beresford, S.A.; Omenn, G.S.; Motulsky, A.G. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057.
- Nygard, O.; Nordrehaug, J.E.; Refsum, H.; Ueland, P.M.; Farstad, M.; Vollset, S.E. Plasma homocysteine levels and mortality in patients with coronary artery disease. N. Engl. J. Med. 1997, 337, 230–236.
- Homocysteine Studies, C. Homocysteine and risk of ischemic heart disease and stroke: A meta-analysis. JAMA 2002, 288, 2015–2022.
- Peng, H.Y.; Man, C.F.; Xu, J.; Fan, Y. Elevated homocysteine levels and risk of cardiovascular and all-cause mortality: A meta-analysis of prospective studies. J. Zhejiang Univ. Sci. B 2015, 16, 78–86.
- den Heijer, M.; Koster, T.; Blom, H.J.; Bos, G.M.; Briet, E.; Reitsma, P.H.; Vandenbroucke, J.P.; Rosendaal, F.R. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N. Engl. J. Med. 1996, 334, 759–762.
- Ray, J.G. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboembolic disease. Arch. Intern. Med. 1998, 158, 2101–2106.
- Ospina-Romero, M.; Cannegieter, S.C.; den Heijer, M.; Doggen, C.J.M.; Rosendaal, F.R.; Lijfering, W.M. Hyperhomocysteinemia and Risk of First Venous Thrombosis: The Influence of (Unmeasured) Confounding Factors. Am. J. Epidemiol. 2018, 187, 1392–1400.
- Cheng, S.W.; Ting, A.C.; Wong, J. Fasting total plasma homocysteine and atherosclerotic peripheral vascular disease. Ann. Vasc. Surg. 1997, 11, 217–223.
- Vasan, R.S.; Beiser, A.; D’Agostino, R.B.; Levy, D.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; Wilson, P.W. Plasma homocysteine and risk for congestive heart failure in adults without prior myocardial infarction. JAMA 2003, 289, 1251–1257.
- Veeranna, V.; Zalawadiya, S.K.; Niraj, A.; Pradhan, J.; Ference, B.; Burack, R.C.; Jacob, S.; Afonso, L. Homocysteine and reclassification of cardiovascular disease risk. J. Am. Coll. Cardiol. 2011, 58, 1025–1033.
- Yuan, D.; Chu, J.; Lin, H.; Zhu, G.; Qian, J.; Yu, Y.; Yao, T.; Ping, F.; Chen, F.; Liu, X. Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 1109445.
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844.
- Nedvetsky, P.I.; Sessa, W.C.; Schmidt, H.H. There’s NO binding like NOS binding: Protein-protein interactions in NO/cGMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 16510–16512.
- Poddar, R.; Sivasubramanian, N.; DiBello, P.M.; Robinson, K.; Jacobsen, D.W. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation 2001, 103, 2717–2723.
- Lei, W.; Long, Y.; Li, S.; Liu, Z.; Zhu, F.; Hou, F.F.; Nie, J. Homocysteine Induces Collagen I Expression by Downregulating Histone Methyltransferase G9a. PLoS ONE 2015, 10, e0130421.
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373.
- Dalton, M.L.; Gadson, P.F., Jr.; Wrenn, R.W.; Rosenquist, T.H. Homocysteine signal cascade: Production of phospholipids, activation of protein kinase C, and the induction of c-fos and c-myb in smooth muscle cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 703–711.
- Nishio, E.; Watanabe, Y. Homocysteine as a modulator of platelet-derived growth factor action in vascular smooth muscle cells: A possible role for hydrogen peroxide. Br. J. Pharmacol. 1997, 122, 269–274.
- Rolland, P.H.; Friggi, A.; Barlatier, A.; Piquet, P.; Latrille, V.; Faye, M.M.; Guillou, J.; Charpiot, P.; Bodard, H.; Ghiringhelli, O.; et al. Hyperhomocysteinemia-induced vascular damage in the minipig. Captopril-hydrochlorothiazide combination prevents elastic alterations. Circulation 1995, 91, 1161–1174.
- Malinow, M.R.; Nieto, F.J.; Szklo, M.; Chambless, L.E.; Bond, G. Carotid artery intimal-medial wall thickening and plasma homocyst(e)ine in asymptomatic adults. The Atherosclerosis Risk in Communities Study. Circulation 1993, 87, 1107–1113.
- Voutilainen, S.; Alfthan, G.; Nyyssonen, K.; Salonen, R.; Salonen, J.T. Association between elevated plasma total homocysteine and increased common carotid artery wall thickness. Ann. Med. 1998, 30, 300–306.
- Arcaro, G.; Fava, C.; Dagradi, R.; Faccini, G.; Gaino, S.; Degan, M.; Lechi, C.; Lechi, A.; Minuz, P. Acute hyperhomocysteinemia induces a reduction in arterial distensibility and compliance. J. Hypertens. 2004, 22, 775–781.
- Upchurch, G.R., Jr.; Welch, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F., Jr.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017.
- Undas, A.; Brozek, J.; Szczeklik, A. Homocysteine and thrombosis: From basic science to clinical evidence. Thromb. Haemost. 2005, 94, 907–915.
- Coppola, A.; Davi, G.; De Stefano, V.; Mancini, F.P.; Cerbone, A.M.; Di Minno, G. Homocysteine, coagulation, platelet function, and thrombosis. Semin. Thromb. Hemost. 2000, 26, 243–254.
- Fryer, R.H.; Wilson, B.D.; Gubler, D.B.; Fitzgerald, L.A.; Rodgers, G.M. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler. Thromb. A J. Vasc. Biol. 1993, 13, 1327–1333.
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J. Clin. Investig. 1991, 88, 1906–1914.
- Rodgers, G.M.; Conn, M.T. Homocysteine, an atherogenic stimulus, reduces protein C activation by arterial and venous endothelial cells. Blood 1990, 75, 895–901.
- Nishinaga, M.; Ozawa, T.; Shimada, K. Homocysteine, a thrombogenic agent, suppresses anticoagulant heparan sulfate expression in cultured porcine aortic endothelial cells. J. Clin. Investig. 1993, 92, 1381–1386.
- Midorikawa, S.; Sanada, H.; Hashimoto, S.; Watanabe, T. Enhancement by homocysteine of plasminogen activator inhibitor-1 gene expression and secretion from vascular endothelial and smooth muscle cells. Biochem. Biophys. Res. Commun. 2000, 272, 182–185.
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14.
- Li, L.; Zhang, Y.; Zeng, C. Update on the epidemiology, genetics, and therapeutic options of hyperuricemia. Am. J. Transl. Res. 2020, 12, 3167–3181.
- Tian, X.; Chen, S.; Zhang, Y.; Zhang, X.; Xu, Q.; Wang, P.; Wu, S.; Luo, Y.; Wang, A. Serum uric acid variation and the risk of cardiovascular disease: A prospective cohort study. Eur. J. Intern. Med. 2023, 112, 37–44.
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680.
- Kanbay, M.; Segal, M.; Afsar, B.; Kang, D.H.; Rodriguez-Iturbe, B.; Johnson, R.J. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart 2013, 99, 759–766.
- Zhang, W.; Iso, H.; Murakami, Y.; Miura, K.; Nagai, M.; Sugiyama, D.; Ueshima, H.; Okamura, T.; Epoch-Japan, G. Serum Uric Acid and Mortality Form Cardiovascular Disease: EPOCH-JAPAN Study. J. Atheroscler. Thromb. 2016, 23, 692–703.
- Maruhashi, T.; Hisatome, I.; Kihara, Y.; Higashi, Y. Hyperuricemia and endothelial function: From molecular background to clinical perspectives. Atherosclerosis 2018, 278, 226–231.
- Kushiyama, A.; Okubo, H.; Sakoda, H.; Kikuchi, T.; Fujishiro, M.; Sato, H.; Kushiyama, S.; Iwashita, M.; Nishimura, F.; Fukushima, T.; et al. Xanthine oxidoreductase is involved in macrophage foam cell formation and atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 291–298.
- Cimmino, G.; Morello, A.; Conte, S.; Pellegrino, G.; Marra, L.; Golino, P.; Cirillo, P. Vitamin D inhibits Tissue Factor and CAMs expression in oxidized low-density lipoproteins-treated human endothelial cells by modulating NF-kappaB pathway. Eur. J. Pharmacol. 2020, 885, 173422.
- Cimmino, G.; Conte, S.; Marra, L.; Morello, A.; Morello, M.; De Rosa, G.; Pepe, M.; Sugralyev, A.; Golino, P.; Cirillo, P. Uric Acid induces a pro-atherothrombotic phenotype in human endothelial cells by imbalancing TF/TFPI pathway. Thromb. Haemost. 2022, 123, 64–75.
- Wang, M.; Lin, X.; Yang, X.; Yang, Y. Research progress on related mechanisms of uric acid activating NLRP3 inflammasome in chronic kidney disease. Ren. Fail. 2022, 44, 615–624.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Cimmino, G.; Gallinoro, E.; di Serafino, L.; De Rosa, G.; Sugraliyev, A.; Golino, P.; Cirillo, P. Uric acid plasma levels are associated with C-reactive protein concentrations and the extent of coronary artery lesions in patients with acute coronary syndromes. Intern. Emerg. Med. 2023.
- Grayson, P.C.; Kim, S.Y.; LaValley, M.; Choi, H.K. Hyperuricemia and incident hypertension: A systematic review and meta-analysis. Arthritis Care Res. 2011, 63, 102–110.
- Tamariz, L.; Agarwal, S.; Soliman, E.Z.; Chamberlain, A.M.; Prineas, R.; Folsom, A.R.; Ambrose, M.; Alonso, A. Association of serum uric acid with incident atrial fibrillation (from the Atherosclerosis Risk in Communities study). Am. J. Cardiol. 2011, 108, 1272–1276.
- Maharani, N.; Kuwabara, M.; Hisatome, I. Hyperuricemia and Atrial Fibrillation. Int. Heart J. 2016, 57, 395–399.
- Holick, M.F. Vitamin D Deficiency. N. Engl. J. Med. 2007, 357, 266–281.
- Macdonald, H.M.; Mavroeidi, A.; Fraser, W.D.; Darling, A.L.; Black, A.J.; Aucott, L.; O’Neill, F.; Hart, K.; Berry, J.L.; Lanham-New, S.A.; et al. Sunlight and dietary contributions to the seasonal vitamin D status of cohorts of healthy postmenopausal women living at northerly latitudes: A major cause for concern? Osteoporos. Int. 2011, 22, 2461–2472.
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, F8–F28.
- Melamed, M.L.; Michos, E.D.; Post, W.; Astor, B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch. Intern. Med. 2008, 168, 1629–1637.
- Kilkkinen, A.; Knekt, P.; Aro, A.; Rissanen, H.; Marniemi, J.; Heliovaara, M.; Impivaara, O.; Reunanen, A. Vitamin D status and the risk of cardiovascular disease death. Am. J. Epidemiol. 2009, 170, 1032–1039.
- Skaaby, T.; Thuesen, B.H.; Linneberg, A. Vitamin D, Cardiovascular Disease and Risk Factors. Adv. Exp. Med. Biol. 2017, 996, 221–230.
- Ginde, A.A.; Scragg, R.; Schwartz, R.S.; Camargo, C.A., Jr. Prospective study of serum 25-hydroxyvitamin D level, cardiovascular disease mortality, and all-cause mortality in older U.S. adults. J. Am. Geriatr. Soc. 2009, 57, 1595–1603.
- Pilz, S.; Dobnig, H.; Nijpels, G.; Heine, R.J.; Stehouwer, C.D.; Snijder, M.B.; van Dam, R.M.; Dekker, J.M. Vitamin D and mortality in older men and women. Clin. Endocrinol. 2009, 71, 666–672.
- Theodoratou, E.; Tzoulaki, I.; Zgaga, L.; Ioannidis, J.P. Vitamin D and multiple health outcomes: Umbrella review of systematic reviews and meta-analyses of observational studies and randomised trials. BMJ 2014, 348, g2035.
- Rostand, S.G. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension 1997, 30, 150–156.
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776.
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238.
- Cimmino, G.; Conte, S.; Morello, M.; Pellegrino, G.; Marra, L.; Morello, A.; Nicoletti, G.; De Rosa, G.; Golino, P.; Cirillo, P. Vitamin D Inhibits IL-6 Pro-Atherothrombotic Effects in Human Endothelial Cells: A Potential Mechanism for Protection against COVID-19 Infection? J. Cardiovasc. Dev. Dis. 2022, 9, 27.
- Bolland, M.J.; Grey, A.; Gamble, G.D.; Reid, I.R. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: A trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014, 2, 307–320.
- Avenell, A.; MacLennan, G.S.; Jenkinson, D.J.; McPherson, G.C.; McDonald, A.M.; Pant, P.R.; Grant, A.M.; Campbell, M.K.; Anderson, F.H.; Cooper, C.; et al. Long-term follow-up for mortality and cancer in a randomized placebo-controlled trial of vitamin D(3) and/or calcium (RECORD trial). J. Clin. Endocrinol. Metab. 2012, 97, 614–622.
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511.
- Tietjens, J.R.; Claman, D.; Kezirian, E.J.; De Marco, T.; Mirzayan, A.; Sadroonri, B.; Goldberg, A.N.; Long, C.; Gerstenfeld, E.P.; Yeghiazarians, Y. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J. Am. Heart Assoc. 2019, 8, e010440.
- Berry, R.B.; Budhiraja, R.; Gottlieb, D.J.; Gozal, D.; Iber, C.; Kapur, V.K.; Marcus, C.L.; Mehra, R.; Parthasarathy, S.; Quan, S.F.; et al. Rules for scoring respiratory events in sleep: Update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2012, 8, 597–619.
- Yeghiazarians, Y.; Jneid, H.; Tietjens, J.R.; Redline, S.; Brown, D.L.; El-Sherif, N.; Mehra, R.; Bozkurt, B.; Ndumele, C.E.; Somers, V.K. Obstructive Sleep Apnea and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2021, 144, e56–e67.
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased prevalence of sleep-disordered breathing in adults. Am. J. Epidemiol. 2013, 177, 1006–1014.
- Johnson, K.G.; Johnson, D.C. Frequency of sleep apnea in stroke and TIA patients: A meta-analysis. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2010, 6, 131–137.
- Worsnop, C.J.; Naughton, M.T.; Barter, C.E.; Morgan, T.O.; Anderson, A.I.; Pierce, R.J. The prevalence of obstructive sleep apnea in hypertensives. Am. J. Respir. Crit. Care Med. 1998, 157, 111–115.
- Fogel, R.B.; Malhotra, A.; White, D.P. Sleep. 2: Pathophysiology of obstructive sleep apnoea/hypopnoea syndrome. Thorax 2004, 59, 159–163.
- Somers, V.K.; Mark, A.L.; Zavala, D.C.; Abboud, F.M. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J. Appl. Physiol. 1989, 67, 2101–2106.
- Somers, V.K.; Dyken, M.E.; Clary, M.P.; Abboud, F.M. Sympathetic neural mechanisms in obstructive sleep apnea. J. Clin. Investig. 1995, 96, 1897–1904.
- Floras, J.S.; Bradley, T.D. Treating obstructive sleep apnea: Is there more to the story than 2 millimeters of mercury? Hypertension 2007, 50, 289–291.
- Hall, M.J.; Ando, S.; Floras, J.S.; Bradley, T.D. Magnitude and time course of hemodynamic responses to Mueller maneuvers in patients with congestive heart failure. J. Appl. Physiol. 1998, 85, 1476–1484.
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective study of the association between sleep-disordered breathing and hypertension. N. Engl. J. Med. 2000, 342, 1378–1384.
- Suzuki, Y.J.; Jain, V.; Park, A.M.; Day, R.M. Oxidative stress and oxidant signaling in obstructive sleep apnea and associated cardiovascular diseases. Free Radic. Biol. Med. 2006, 40, 1683–1692.
- Ambrosio, G.; Tritto, I. Reperfusion injury: Experimental evidence and clinical implications. Am. Heart J. 1999, 138, S69–S75.
- Ambrosio, G.; Zweier, J.L.; Duilio, C.; Kuppusamy, P.; Santoro, G.; Elia, P.P.; Tritto, I.; Cirillo, P.; Condorelli, M.; Chiariello, M. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 1993, 268, 18532–18541.
- Schulz, R.; Schmidt, D.; Blum, A.; Lopes-Ribeiro, X.; Lucke, C.; Mayer, K.; Olschewski, H.; Seeger, W.; Grimminger, F. Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea: Response to CPAP therapy. Thorax 2000, 55, 1046–1051.
- Schulz, R.; Seeger, W.; Grimminger, F. Serum nitrite/nitrate levels in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2001, 164, 1997–1998.
- Logan, A.G.; Perlikowski, S.M.; Mente, A.; Tisler, A.; Tkacova, R.; Niroumand, M.; Leung, R.S.; Bradley, T.D. High prevalence of unrecognized sleep apnoea in drug-resistant hypertension. J. Hypertens. 2001, 19, 2271–2277.
- Hoffstein, V.; Mateika, J. Evening-to-morning blood pressure variations in snoring patients with and without obstructive sleep apnea. Chest 1992, 101, 379–384.
- Leung, R.S.; Bradley, T.D. Sleep apnea and cardiovascular disease. Am. J. Respir. Crit. Care Med. 2001, 164, 2147–2165.
- Loredo, J.S.; Ancoli-Israel, S.; Dimsdale, J.E. Sleep quality and blood pressure dipping in obstructive sleep apnea. Am. J. Hypertens. 2001, 14, 887–892.
- Portaluppi, F.; Provini, F.; Cortelli, P.; Plazzi, G.; Bertozzi, N.; Manfredini, R.; Fersini, C.; Lugaresi, E. Undiagnosed sleep-disordered breathing among male nondippers with essential hypertension. J. Hypertens. 1997, 15, 1227–1233.
- Liu, L.; Cao, Q.; Guo, Z.; Dai, Q. Continuous Positive Airway Pressure in Patients with Obstructive Sleep Apnea and Resistant Hypertension: A Meta-Analysis of Randomized Controlled Trials. J. Clin. Hypertens. 2016, 18, 153–158.
- Patel, N.; Donahue, C.; Shenoy, A.; Patel, A.; El-Sherif, N. Obstructive sleep apnea and arrhythmia: A systemic review. Int. J. Cardiol. 2017, 228, 967–970.
- Roche, F.; Xuong, A.N.; Court-Fortune, I.; Costes, F.; Pichot, V.; Duverney, D.; Vergnon, J.M.; Gaspoz, J.M.; Barthelemy, J.C. Relationship among the severity of sleep apnea syndrome, cardiac arrhythmias, and autonomic imbalance. Pacing Clin. Electrophysiol. PACE 2003, 26, 669–677.
- Shamsuzzaman, A.S.; Winnicki, M.; Lanfranchi, P.; Wolk, R.; Kara, T.; Accurso, V.; Somers, V.K. Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 2002, 105, 2462–2464.
- Kuniyoshi, F.H.; Garcia-Touchard, A.; Gami, A.S.; Romero-Corral, A.; van der Walt, C.; Pusalavidyasagar, S.; Kara, T.; Caples, S.M.; Pressman, G.S.; Vasquez, E.C.; et al. Day-night variation of acute myocardial infarction in obstructive sleep apnea. J. Am. Coll. Cardiol. 2008, 52, 343–346.
- Zhao, Q.; Liu, Z.H.; Zhao, Z.H.; Luo, Q.; McEvoy, R.D.; Zhang, H.L.; Wang, Y. Effects of obstructive sleep apnea and its treatment on cardiovascular risk in CAD patients. Respir. Med. 2011, 105, 1557–1564.
- Brown, D.L.; Shafie-Khorassani, F.; Kim, S.; Chervin, R.D.; Case, E.; Morgenstern, L.B.; Yadollahi, A.; Tower, S.; Lisabeth, L.D. Sleep-Disordered Breathing Is Associated with Recurrent Ischemic Stroke. Stroke 2019, 50, 571–576.
- Lisabeth, L.D.; Sanchez, B.N.; Lim, D.; Chervin, R.D.; Case, E.; Morgenstern, L.B.; Tower, S.; Brown, D.L. Sleep-disordered breathing and poststroke outcomes. Ann. Neurol. 2019, 86, 241–250.
- Kholdani, C.; Fares, W.H.; Mohsenin, V. Pulmonary hypertension in obstructive sleep apnea: Is it clinically significant? A critical analysis of the association and pathophysiology. Pulm. Circ. 2015, 5, 220–227.
- Oldenburg, O.; Lamp, B.; Faber, L.; Teschler, H.; Horstkotte, D.; Topfer, V. Sleep-disordered breathing in patients with symptomatic heart failure: A contemporary study of prevalence in and characteristics of 700 patients. Eur. J. Heart Fail. 2007, 9, 251–257.
- Phillips, B.G.; Kato, M.; Narkiewicz, K.; Choe, I.; Somers, V.K. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H234–H237.
- Wolk, R.; Somers, V.K. Sleep and the metabolic syndrome. Exp. Physiol. 2007, 92, 67–78.
- Vgontzas, A.N.; Papanicolaou, D.A.; Bixler, E.O.; Hopper, K.; Lotsikas, A.; Lin, H.M.; Kales, A.; Chrousos, G.P. Sleep apnea and daytime sleepiness and fatigue: Relation to visceral obesity, insulin resistance, and hypercytokinemia. J. Clin. Endocrinol. Metab. 2000, 85, 1151–1158.
- Perez Velasco, R.; Jarosinska, D. Update of the WHO global air quality guidelines: Systematic reviews—An introduction. Environ. Int. 2022, 170, 107556.
- Collaborators, G.B.D.R.F. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249.
- Liu, C.; Chen, R.; Sera, F.; Vicedo-Cabrera, A.M.; Guo, Y.; Tong, S.; Coelho, M.; Saldiva, P.H.N.; Lavigne, E.; Matus, P.; et al. Ambient Particulate Air Pollution and Daily Mortality in 652 Cities. N. Engl. J. Med. 2019, 381, 705–715.
- Yusuf, S.; Joseph, P.; Rangarajan, S.; Islam, S.; Mente, A.; Hystad, P.; Brauer, M.; Kutty, V.R.; Gupta, R.; Wielgosz, A.; et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): A prospective cohort study. Lancet 2020, 395, 795–808.
- Kaufman, J.D.; Adar, S.D.; Barr, R.G.; Budoff, M.; Burke, G.L.; Curl, C.L.; Daviglus, M.L.; Diez Roux, A.V.; Gassett, A.J.; Jacobs, D.R., Jr.; et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): A longitudinal cohort study. Lancet 2016, 388, 696–704.
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378.
- Newby, D.E.; Mannucci, P.M.; Tell, G.S.; Baccarelli, A.A.; Brook, R.D.; Donaldson, K.; Forastiere, F.; Franchini, M.; Franco, O.H.; Graham, I.; et al. Expert position paper on air pollution and cardiovascular disease. Eur. Heart J. 2015, 36, 83–93.
- Rajagopalan, S.; Al-Kindi, S.G.; Brook, R.D. Air Pollution and Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2054–2070.
- Shah, A.S.; Lee, K.K.; McAllister, D.A.; Hunter, A.; Nair, H.; Whiteley, W.; Langrish, J.P.; Newby, D.E.; Mills, N.L. Short term exposure to air pollution and stroke: Systematic review and meta-analysis. BMJ 2015, 350, h1295.
- Schraufnagel, D.E.; Balmes, J.R.; Cowl, C.T.; De Matteis, S.; Jung, S.H.; Mortimer, K.; Perez-Padilla, R.; Rice, M.B.; Riojas-Rodriguez, H.; Sood, A.; et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 2: Air Pollution and Organ Systems. Chest 2019, 155, 417–426.
- Stafoggia, M.; Cesaroni, G.; Peters, A.; Andersen, Z.J.; Badaloni, C.; Beelen, R.; Caracciolo, B.; Cyrys, J.; de Faire, U.; de Hoogh, K.; et al. Long-term exposure to ambient air pollution and incidence of cerebrovascular events: Results from 11 European cohorts within the ESCAPE project. Environ. Health Perspect. 2014, 122, 919–925.
- Baldauf, R.W.; Devlin, R.B.; Gehr, P.; Giannelli, R.; Hassett-Sipple, B.; Jung, H.; Martini, G.; McDonald, J.; Sacks, J.D.; Walker, K. Ultrafine Particle Metrics and Research Considerations: Review of the 2015 UFP Workshop. Int. J. Environ. Res. Public Health 2016, 13, 1054.
- Shukla, A.; Timblin, C.; BeruBe, K.; Gordon, T.; McKinney, W.; Driscoll, K.; Vacek, P.; Mossman, B.T. Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am. J. Respir. Cell Mol. Biol. 2000, 23, 182–187.
- Roy, A.; Gong, J.; Thomas, D.C.; Zhang, J.; Kipen, H.M.; Rich, D.Q.; Zhu, T.; Huang, W.; Hu, M.; Wang, G.; et al. The cardiopulmonary effects of ambient air pollution and mechanistic pathways: A comparative hierarchical pathway analysis. PLoS ONE 2014, 9, e114913.
- Daiber, A.; Oelze, M.; Steven, S.; Kroller-Schon, S.; Munzel, T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol. 2017, 12, 35–49.
- Haberzettl, P.; Lee, J.; Duggineni, D.; McCracken, J.; Bolanowski, D.; O’Toole, T.E.; Bhatnagar, A.; Conklin, D.J. Exposure to ambient air fine particulate matter prevents VEGF-induced mobilization of endothelial progenitor cells from the bone marrow. Environ. Health Perspect. 2012, 120, 848–856.
- Nemmar, A.; Vanbilloen, H.; Hoylaerts, M.F.; Hoet, P.H.; Verbruggen, A.; Nemery, B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am. J. Respir. Crit. Care Med. 2001, 164, 1665–1668.
- Kreyling, W.G.; Semmler, M.; Erbe, F.; Mayer, P.; Takenaka, S.; Schulz, H.; Oberdorster, G.; Ziesenis, A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J. Toxicol. Environ. Health Part A 2002, 65, 1513–1530.
- Oberdorster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Lunts, A.; Kreyling, W.; Cox, C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J. Toxicol. Environ. Health Part A 2002, 65, 1531–1543.
- Miller, M.R.; Raftis, J.B.; Langrish, J.P.; McLean, S.G.; Samutrtai, P.; Connell, S.P.; Wilson, S.; Vesey, A.T.; Fokkens, P.H.B.; Boere, A.J.F.; et al. Inhaled Nanoparticles Accumulate at Sites of Vascular Disease. ACS Nano 2017, 11, 4542–4552.
- Nemmar, A.; Hoet, P.H.; Dinsdale, D.; Vermylen, J.; Hoylaerts, M.F.; Nemery, B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation 2003, 107, 1202–1208.
- Jacobs, L.; Emmerechts, J.; Mathieu, C.; Hoylaerts, M.F.; Fierens, F.; Hoet, P.H.; Nemery, B.; Nawrot, T.S. Air pollution related prothrombotic changes in persons with diabetes. Environ. Health Perspect. 2010, 118, 191–196.
- Ying, Z.; Xu, X.; Bai, Y.; Zhong, J.; Chen, M.; Liang, Y.; Zhao, J.; Liu, D.; Morishita, M.; Sun, Q.; et al. Long-term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: A role for hypothalamic inflammation. Environ. Health Perspect. 2014, 122, 79–86.
- Bartoli, C.R.; Wellenius, G.A.; Coull, B.A.; Akiyama, I.; Diaz, E.A.; Lawrence, J.; Okabe, K.; Verrier, R.L.; Godleski, J.J. Concentrated ambient particles alter myocardial blood flow during acute ischemia in conscious canines. Environ. Health Perspect. 2009, 117, 333–337.
- Niu, Z.; Liu, F.; Li, B.; Li, N.; Yu, H.; Wang, Y.; Tang, H.; Chen, X.; Lu, Y.; Cheng, Z.; et al. Acute effect of ambient fine particulate matter on heart rate variability: An updated systematic review and meta-analysis of panel studies. Environ. Health Prev. Med. 2020, 25, 77.
- Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: Standards of measurement, physiological interpretation and clinical use. Circulation 1996, 93, 1043–1065.
- Mustafic, H.; Jabre, P.; Caussin, C.; Murad, M.H.; Escolano, S.; Tafflet, M.; Perier, M.C.; Marijon, E.; Vernerey, D.; Empana, J.P.; et al. Main air pollutants and myocardial infarction: A systematic review and meta-analysis. JAMA 2012, 307, 713–721.
- Pope, C.A.; Muhlestein, J.B.; Anderson, J.L.; Cannon, J.B.; Hales, N.M.; Meredith, K.G.; Le, V.; Horne, B.D. Short-Term Exposure to Fine Particulate Matter Air Pollution Is Preferentially Associated with the Risk of ST-Segment Elevation Acute Coronary Events. J. Am. Heart Assoc. 2015, 4, e002506.
- Baneras, J.; Ferreira-Gonzalez, I.; Marsal, J.R.; Barrabes, J.A.; Ribera, A.; Lidon, R.M.; Domingo, E.; Marti, G.; Garcia-Dorado, D.; Codi, I.A.M.R.i. Short-term exposure to air pollutants increases the risk of ST elevation myocardial infarction and of infarct-related ventricular arrhythmias and mortality. Int. J. Cardiol. 2018, 250, 35–42.
- Cesaroni, G.; Forastiere, F.; Stafoggia, M.; Andersen, Z.J.; Badaloni, C.; Beelen, R.; Caracciolo, B.; de Faire, U.; Erbel, R.; Eriksen, K.T.; et al. Long term exposure to ambient air pollution and incidence of acute coronary events: Prospective cohort study and meta-analysis in 11 European cohorts from the ESCAPE Project. BMJ 2014, 348, f7412.
- Jilani, M.H.; Simon-Friedt, B.; Yahya, T.; Khan, A.Y.; Hassan, S.Z.; Kash, B.; Blankstein, R.; Blaha, M.J.; Virani, S.S.; Rajagopalan, S.; et al. Associations between particulate matter air pollution, presence and progression of subclinical coronary and carotid atherosclerosis: A systematic review. Atherosclerosis 2020, 306, 22–32.
- Provost, E.B.; Madhloum, N.; Int Panis, L.; De Boever, P.; Nawrot, T.S. Carotid intima-media thickness, a marker of subclinical atherosclerosis, and particulate air pollution exposure: The meta-analytical evidence. PLoS ONE 2015, 10, e0127014.
- Khraishah, H.; Alahmad, B.; Ostergard, R.L., Jr.; AlAshqar, A.; Albaghdadi, M.; Vellanki, N.; Chowdhury, M.M.; Al-Kindi, S.G.; Zanobetti, A.; Gasparrini, A.; et al. Climate change and cardiovascular disease: Implications for global health. Nat. Rev. Cardiol. 2022, 19, 798–812.
- Jacobsen, A.P.; Khiew, Y.C.; Duffy, E.; O’Connell, J.; Brown, E.; Auwaerter, P.G.; Blumenthal, R.S.; Schwartz, B.S.; McEvoy, J.W. Climate change and the prevention of cardiovascular disease. Am. J. Prev. Cardiol. 2022, 12, 100391.
- Alahmad, B.; Khraishah, H.; Royé, D.; Vicedo-Cabrera, A.M.; Guo, Y.; Papatheodorou, S.I.; Achilleos, S.; Acquaotta, F.; Armstrong, B.; Bell, M.L.; et al. Associations Between Extreme Temperatures and Cardiovascular Cause-Specific Mortality: Results From 27 Countries. Circulation 2023, 147, 35–46.
- Kysely, J.; Pokorna, L.; Kyncl, J.; Kriz, B. Excess cardiovascular mortality associated with cold spells in the Czech Republic. BMC Public Health 2009, 9, 19.
- Weerasinghe, D.P.; MacIntyre, C.R.; Rubin, G.L. Seasonality of coronary artery deaths in New South Wales, Australia. Heart 2002, 88, 30–34.
- Rogot, E.; Padgett, S.J. Associations of coronary and stroke mortality with temperature and snowfall in selected areas of the United States, 1962–1966. Am. J. Epidemiol. 1976, 103, 565–575.
- Kan, H.D.; Jia, J.; Chen, B.H. Temperature and daily mortality in Shanghai: A time-series study. Biomed. Environ. Sci. BES 2003, 16, 133–139.
- De Lorenzo, F.; Kadziola, Z.; Mukherjee, M.; Saba, N.; Kakkar, V.V. Haemodynamic responses and changes of haemostatic risk factors in cold-adapted humans. QJM Mon. J. Assoc. Physicians 1999, 92, 509–513.
- Neild, P.J.; Syndercombe-Court, D.; Keatinge, W.R.; Donaldson, G.C.; Mattock, M.; Caunce, M. Cold-induced increases in erythrocyte count, plasma cholesterol and plasma fibrinogen of elderly people without a comparable rise in protein C or factor X. Clin. Sci. 1994, 86, 43–48.
- Gasparrini, A.; Guo, Y.; Hashizume, M.; Lavigne, E.; Zanobetti, A.; Schwartz, J.; Tobias, A.; Tong, S.; Rocklov, J.; Forsberg, B.; et al. Mortality risk attributable to high and low ambient temperature: A multicountry observational study. Lancet 2015, 386, 369–375.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
861
Revisions:
3 times
(View History)
Update Date:
30 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No