Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniela Damiani | -- | 3776 | 2023-08-28 15:56:35 | | | |
| 2 | Rita Xu | Meta information modification | 3776 | 2023-08-29 04:59:31 | | | | |
| 3 | Rita Xu | Meta information modification | 3776 | 2023-08-30 09:40:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Damiani, D.; Tiribelli, M. Checkpoint Inhibitors in Acute Myeloid Leukemia. Encyclopedia. Available online: https://encyclopedia.pub/entry/48545 (accessed on 09 August 2026).
Damiani D, Tiribelli M. Checkpoint Inhibitors in Acute Myeloid Leukemia. Encyclopedia. Available at: https://encyclopedia.pub/entry/48545. Accessed August 09, 2026.
Damiani, Daniela, Mario Tiribelli. "Checkpoint Inhibitors in Acute Myeloid Leukemia" Encyclopedia, https://encyclopedia.pub/entry/48545 (accessed August 09, 2026).
Damiani, D., & Tiribelli, M. (2023, August 28). Checkpoint Inhibitors in Acute Myeloid Leukemia. In Encyclopedia. https://encyclopedia.pub/entry/48545
Damiani, Daniela and Mario Tiribelli. "Checkpoint Inhibitors in Acute Myeloid Leukemia." Encyclopedia. Web. 28 August, 2023.
Copy Citation
The prognosis of acute myeloid leukemia (AML) remains unsatisfactory. Among the reasons for the poor response to therapy and high incidence of relapse, there is tumor cell immune escape, as AML blasts can negatively influence various components of the immune system, mostly weakening T-cells.
acute myeloid leukemia
immune microenvironment
checkpoint inhibitors
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease in which conventional treatment is still associated with unacceptably high rates of relapse and death despite complete remission (CR) being obtained in around 70% of patients [1]. The disease results from the transformation of rapidly proliferating progenitor cells. In AML, dysfunctional hierarchy prevails over the normal one, resulting in progressive cytopenias and increased risks of bleeding and infections.
In recent years, much progress has been made concerning the knowledge of the molecular pathogenesis of AML [2][3], and new drugs acting on various genetic abnormalities have been developed, thus permitting novel therapeutic approaches and, eventually, higher survival rates [4]. The treatment strategies used for AML depend on many factors, both disease-specific (e.g., prior exposure to toxics, prior myelodysplastic syndrome—MDS—cytogenetic and molecular abnormalities, and relapsed or refractory disease) and patient-specific (e.g., age, comorbidities, and previous exposure to chemotherapy). The current treatment for most cases of AML, excluding acute promyelocytic leukemia, consists of two phases: CR induction and the consolidation phase. In patients eligible for intensive induction, the “classical” 3 + 7 regimen is generally used; alternatives include the addition of fludarabine (FLAI) or mitoxantrone-based regimens [5]. In addition, the use of the FLT3 inhibitor midostaurin has become a standard in AML patients harboring FLT3-ITD mutation [5][6]. In patients not eligible for intensive chemotherapy, hypomethylating agents (HMAs), alone or in association with the BCL2 inhibitor venetoclax or IDH1 inhibitor ivosidenib, are the most recent options [5]. In “fit” patients, consolidation includes intermediate or high-dose cytarabine and allogeneic hematopoietic cell transplantation (HCT), which is still considered the only curative option [7]. However, the “cure” of AML remains a major challenge, as almost half of transplanted patients relapse after HCT, a risk that depends on disease status at transplant, donor type, T cell depletion strategies, and conditioning regimen [8]. It has now become evident that AML development and progression, as well as drug resistance acquisition, are associated with changes in the bone marrow microenvironment, which provide physical protection and release pro-survival factors for leukemic cells and induce dysregulated immune responses that facilitate the immune escape of AML blasts [9]. In addition to targeting molecular alterations, the manipulation of the AML immune microenvironment to restore immune surveillance appears to be an attractive strategy.
2. Checkpoint Inhibitors in AML Therapy
The evidence that immune checkpoint interactions contribute to AML immune evasion, not only at relapse but also in newly diagnosed disease, represents the rationale to use this therapeutic approach to reactivate immune sensitivity through a block of co-inhibitory ligands. From the approval of the first antibody blocking an immune checkpoint in 2011, immune checkpoint inhibitors (ICI) revolutionized the oncology field, resulting in durable response rates in patients with historically limited therapeutic options [10]. After approval in solid malignancies, various molecules, such as ipilimumab, pembrolizumab, cemiplimab, avelumab, and durvalumab, acting on specific targets, are undergoing clinical trials in relation to AML [11]. Due to their limited activity when used as a monotherapy, despite the demonstrated ability of co-inhibitory blockade and improved immune response, combination trials are ongoing. Despite the promising activity of checkpoint inhibitors in solid tumors [12], so far, clinical trials relating to AML have reported controversial results, possibly due to the low mutational burden and the DNA mismatch repair proficiency in AML when compared to solid tumors [13][14]. However, about fifty trials are currently ongoing testing ICI in AML and MDS, either as monotherapy or in combination with chemotherapy or HMAs. Key ICI potential targets in AML are shown in Figure 1.
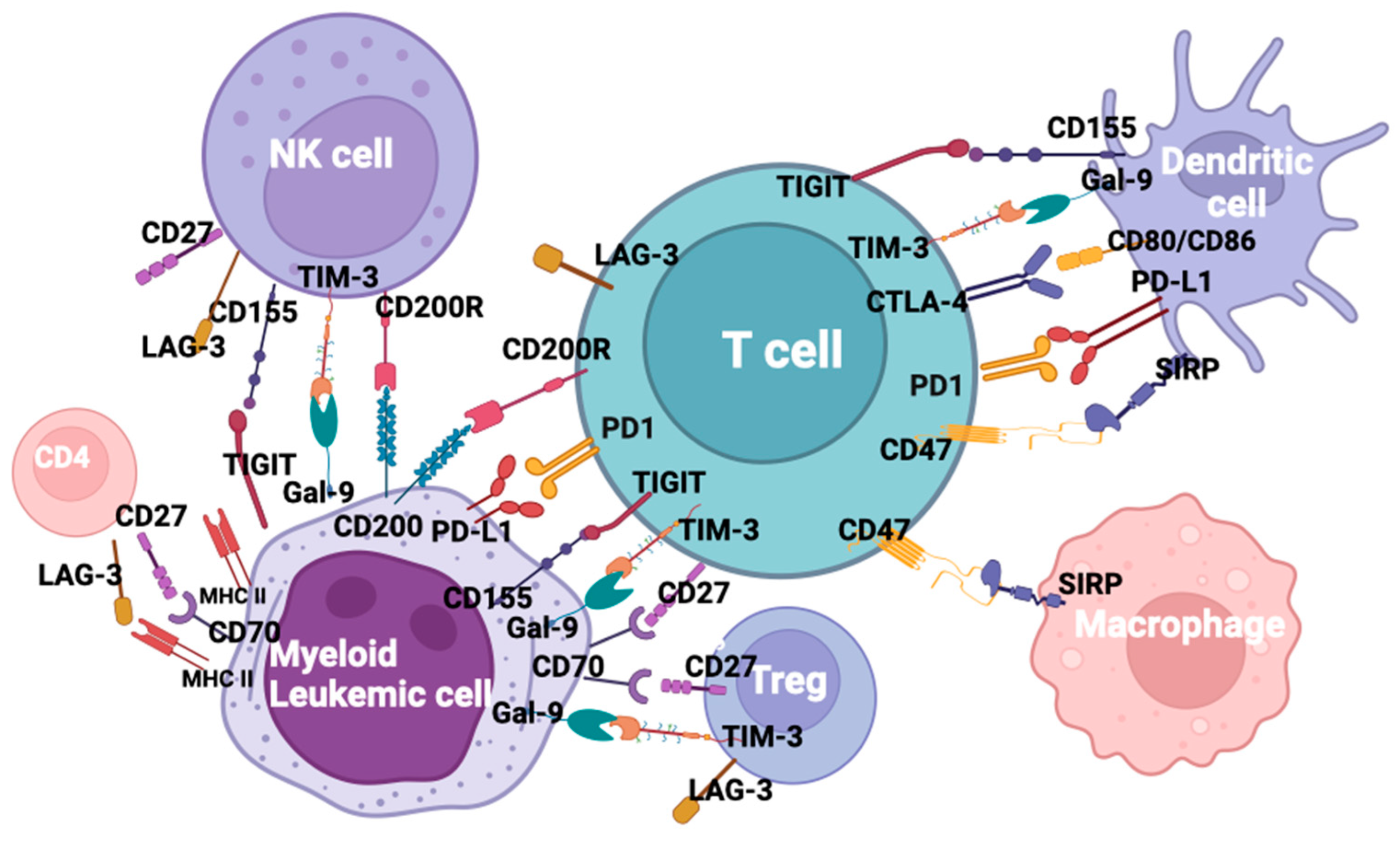
Figure 1. Checkpoint inhibitors and ligands in acute myeloid leukemia.
2.1. The CTLA-4/B7 Axis
CTLA-4 (CD152) is a member of the immunoglobulin-related receptors interacting with CD80 and CD86 ligands to deliver an inhibitory signal aimed at terminating immune responses. Moreover, it has a role in Tregs induction and, consequently, in regulating tolerance and autoimmunity [15]. The aberrant expression of CTLA-4 has been reported in AML, with a detrimental effect on disease outcomes [16]. In animal models, blocking CTLA-4 enhanced T-cell activity and suppressed Tregs [17]. In clinical trials relating to melanoma, anti-CTLA-4 ipilimumab increased the Teff/Tregs ratio, enhanced NK activity and restored T-effector function, ultimately prolonging survival [18][19]. Data on AML cell lines and preliminary results from clinical trials confirmed what has been reported in solid tumors [20]. Zhong et al. have shown that anti-CTLA-4 can improve AML-specific T-cell function in terms of frequency, cytotoxic capacity and IFN-γ secretion [21]. Data on the “in vivo” efficacy of ipilimumab are scarce. Davids and coll. reported rates of CR and partial response (PR) of 23% and 9%, respectively, in hematological malignancies receiving single-agent ipilimumab for relapse after HCT [22]. Bashey et al. observed no responses in AML patients among a small cohort of 29 patients with various hematologic malignancies relapsed after HCT [23]. A phase I trial of ipilimumab and a definite dose of Treg-depleted donor lymphocyte infusion (DLI) for AML and MDS relapsed after HCT is recruited (NCT03912064).
HMAs have been shown to upregulate checkpoint receptors and their ligands in AML/MDS models [24]. The “in vitro” data correlates to a clinical study in AML, showing that the azacytidine (AZA)-induced demethylation of PD-1 promoter translates into the increased expression of PD-1 and an unfavorable outcome in the absence of immunotherapy [25]. A phase I trial combining ipilimumab and decitabine (DAC) in relapsed/refractory AML and MDS after allogeneic HCT or HCT-naïve is currently active but not recruiting (NCT2890329).
2.2. The PD1/PD-L1 Axis
Programmed death receptor-1 (PD1 or CD279) is a type I transmembrane protein expressed mostly in activated immune cells [26]. PD1 binds two ligands: PD-L1 (CD274) and PD-L2 (CD273). PD-L1 is a member of the B7 family of co-stimulatory/co-inhibitory molecules present in hematopoietic cells and is upregulated or aberrantly expressed in many tumors [26][27]. In AML, the up-regulation of PD1 was reported in T-effector cells and in Tregs [28]; it is hypothesized that PD1 overexpression on CD8+ T-cells may account for CTL dysfunction and immune suppression during AML progression [29]. Moreover, PD1 and T-cell immunoglobulin and mucin domain 3 (TIM-3) co-expression on T-cells has been correlated to T-cell exhaustion in human and murine models [30] and can predict leukemia relapse post-HCT [31]. In addition, the co-expression of PD1 and TIM-3 seems to be more frequent in patients experiencing multiple relapses [28]. On the other hand, the up-regulation of PD-L1 and PD-L2 at diagnosis, relapse and during treatment correlates with resistance to therapy and poor prognosis [32][33][34][35]. PD1/PD-L1 engagement drives an inhibitory signal causing T-cell exhaustion and favoring the immune escape of neoplastic cells [26]. Moreover, it induces the apoptosis of tumor-specific cells and favors Tregs differentiation and resistance to CD8+ mediated cytolysis [36].
Three PD1-blocking antibodies (nivolumab, pembrolizumab and cemiplimab) and three PD-L1 inhibitors (atezolizumab, avelumab and durvalumab) have been approved to date by the FDA for various solid tumors [11]. However, PD1 and PD-L1 inhibitors have still not been approved for AML.
The results of a few clinical trials of drugs against the PD1–PD-L1 axis that have been completed or are ongoing in relation to AML are summarized in Table 1. Nivolumab was used alone as maintenance in high-risk AML [37] or to eliminate MRD. An ongoing phase II trial is exploring the tolerability of nivolumab in relapsed AML or MRD-positive AML after HCT (NCT03146468), while a phase I/Ib was terminated due to early toxicity. Additionally, pembrolizumab has been evaluated as a maintenance therapy in elderly AML patients or as a salvage therapy after relapse post-allogeneic transplant.
Many clinical trials combining epigenetic therapies with ICI are currently recruiting (NCT03969446, NCT02996474, NCT4277442) [38]. However, patients who received prior HMA therapy displayed lower ORR compared to HMA-naïve patients (22% vs. 58%), suggesting that the combination of HMA and ICI may have better efficacy when administered early in the course of the disease. Daver et al. presented the interim results of the NCT02397720 trial, in which patients with relapsed/refractory AML or elderly patients with newly diagnosed AML were parallel assigned to receive AZA + nivolumab or AZA + nivolumab + ipilimumab: in the latter cohort, ORR and CR/PR rates were 44% and 36%, respectively. The combination of AZA and pembrolizumab in relapsed/refractory AML and in newly diagnosed AML patients over 65 years was investigated in the phase II trial NCT02845297, showing modest clinical activity [39].
Another phase II trial of nivolumab, cytarabine and idarubicin in newly diagnosed AML and MDS (NCT02464657) reported that responders had no evidence of MRD at the time of response, and non-responders were more likely to have TP 53 mutations and a higher frequency of BM CD4+ cells co-expressing PD1 and TIM-3 [40]. The phase II NCT02768792 trial is evaluating the efficacy of the association of pembrolizumab and high-dose cytarabine (HiDAC) in relapsed/refractory AML. The preliminary results showed that pretreatment CD8+ TCR diversity was associated with response, as well as increased chemokine receptors in peripheral blood CD8+ cells and the increased expression of genes involved in p53, IFN-γ and Il-6 pathways [41]. No trial is currently ongoing to test cemiplimab in AMLs.
The preliminary results of the combination of AZA and durvalumab in MDS and AML, which are unfit for intensive chemotherapy, showed an ORR of 61.9% in MDS and of 31% in AML and median OS 11.6 months in MDS and 13.0 months in AML (NCT02775903) [42]. Many phases I and II trials investigating the PD-L1 inhibitors avelumab or atezolizumab in association with HMAs (AZA, DAC or guadecitabine) are recruiting (NCT02953561, NCT03395973, NCT02935361, NCT02892318, NCT3154827), but no results are available at present.
Table 1. Summary of results of a few clinical trials of drugs against PD1–PD-L1 axis completed or ongoing in AML.
| Trial Identifier | Drug | Disease | Response | Survival | Ref. |
|---|---|---|---|---|---|
| NCT02532231 | Nivolumab | HR AML | CR 79% at 6 months, 71% at 12 months |
OS 86% at 12 mo., 67% at 18 mo. |
[37] |
| NCT02397720 | Nivolumab + AZA | RR AML | ORR 58%, CR/CRi 21%, HI 7% | Median OS 9.2 mo. | [38] |
| AZA-naïve AML | ORR 22% | ||||
| NCT02464657 | Nivolumab + cytarabine + idarubicin | Newly diagnosed AML and MDS | CR/CRi 80% | Median OS 18.5 mo. | [40] |
| NCT02845297 | Pembrolizumab + AZA | RR AML | ORR 32%, CR/CRi 14% | Median OS 10.8 mo. | [39] |
| Newly diagnosed elderly AML | CR/CRi 47% | Median OS 13.1 mo. | |||
| NCT02768792 | Pembrolizumab + cytarabine | RR AML | ORR 46%, CR/CRi 38% | [41] | |
| NCT02775903 | Durvalumab + AZA | Unfit MDS and AML | ORR 61.9% in MDS ORR 31% in AML |
Median OS 11.6 mo. MDS Median OS 13 mo. AML |
[42] |
2.3. The TIM-3/Galectin9 Axis
TIM-3 is a co-inhibitory receptor expressed on CD4+ Th1 cells, CD8+ cytotoxic T-cells (CTLs) and other cells of innate immunity, such as dendritic cells, monocytes, macrophages, mast cells and NK cells, as well as on different neoplastic cells [43][44][45]. TIM-3 gene is coded on chromosome 5q33.2, in the same region of IL4 and IL-5 genes. TIM-3 is a single transmembrane (TM) molecule whose extracellular tail contains an N-terminal IgV domain, followed by a mucin domain with glycosylation sites. After this, there is the TM domain and the cytoplasmic tail in the C-terminus. TIM-3 does not contain classic inhibitory tyrosine-based motifs; however, there is a conserved region with five tyrosine residues that are phosphorylated after the interaction of TIM-3 with their ligands [46]. Among the four known ligands of TIM-3, the first and most studied is galectin-9 (gal-9), which induces the apoptosis of Th1 cells [47], playing a crucial role in tumor cell immune evasion. TIM-3 overexpression in human and murine tumor models results in T-cell dysfunction [48]. TIM-3 is often co-expressed with PD-1, and blocking TIM-3 alone or with other co-inhibitory molecules reverse T-cell exhaustion [35]. In AML, high levels of TIM-3 have been found in immune cells, particularly T-cells and NK cells, promoting immune exhaustion, and on LSCs, where it represents a distinctive marker. The overexpression of TIM-3 has been described in LSCs but not in healthy hematopoietic stem cells [49][50]. Kikushige et al. suggested that, on LSCs, TIM-3 and its ligand create an autocrine loop, leading to the phosphorylation of ERK and AKT. This process results in the induction of the β-catenin pathway and NF-kB activation, which regulate development and self-survival [51]. TIM-3 is involved in immune evasion through different mechanisms. Folgiero et al. established that the production of indoleamine 2,3-dioxigenase 1 (IDO1), an anti-inflammatory enzyme, can be stimulated by the IFN-γ released by NK cells after TIM-3/Gal-9 binding [52]. Goncalves Silva et al. reported that the soluble form of TIM-3, formed by its shedding from the surface of AML blasts, inhibits the release of interleukin2 (IL-2), involved in the activation and function of T- and NK cells [53]. Its high expression on immune cells has been associated with a worse prognosis in solid and hematologic neoplasms [26][35]. Li et al. found higher TIM-3 overexpression in CD4+ T-cells from patients with FLT3-mutated compared to non-mutated AML, and in CD8+ cells of high-risk compared to low-risk AML patients [54]. Kong et al. reported shorter leukemia-free survival (LFS) after HCT in patients with high numbers of TIM-3/PD-1 co-expressing T-cells [31]. Zahran et al. demonstrated the association between TIM-3 upregulation and poor prognosis in AML with normal cytogenetics [55]. Tan et al. reported that the overexpression of TIM-3 on CD8+ cells was associated with a negative prognosis [56]. Less defined is the prognostic role of TIM-3 expression in leukemic cells: Tan et al. observed the highest TIM-3 level in M4 AMLs [57]. Dama et al. correlated TIM-3 expression on blasts and chemotherapy failure [58], while Xu et al. reported a better response to chemotherapy in cases with TIM-3-expressing blasts [59]. More studies are needed to clarify the relationship between the expression of TIM-3 on AML blast cells and response to chemotherapy and, which is most important, the mechanisms inducing the up-regulation of TIM-3 on LCSs and its possible impact on the biology of LSCs.
However, taken together, the available data points to TIM-3 as an ideal candidate for therapy with monoclonal antibodies (MoAb). The inhibition of TIM-3/gal-9 binding in “vitro” reduced the proliferation capacity of AML cells [60], and, in murine models, an anti-human TIM-3 MoAb eliminated LSCs without affecting normal hematopoiesis [51]. At present, several MoAbs are being tested in trials for solid tumors, such as MBG453 (sabatolimab), TSR-022, BMS-986258, LY3321367, SYM023, BGB-A425, SHR 1702 [61], but only MBG453 has shown preliminary efficacy and safety in AML and MDS. Currently, eight phase I/II trials of MBG453 in monotherapy or in combination with HMAs, PD-1 inhibitors, the MDM2 inhibitor HDM201, and venetoclax are ongoing. The interim data from the NCT03066648 trial (MBG453 + HMAs) reported an ORR of 58% in MDS and 38% in newly diagnosed AML patients for the MBG453 + DAC arm and 70% in MDS and 27% in AML patients for the MBG453 + AZA arm, respectively. The commonest grade 3/4 AEs were thrombocytopenia, anemia and neutropenia (febrile or not); in the MBG453 + DAC group, four immune-related events were reported (ALT increase, arthritis, hepatitis and hypothyroidism), compared to none in the MBG453 + AZA cohort [62][63].
2.4. The LAG-3/MHC Axis
LAG-3 (CD223) is a CD4-like molecule binding MHC class II with a greater affinity than CD4, generating a signal blocking T-cell activation [18]. The protein is coded by a gene in the short arm of chromosome 12 (12p13.32) in humans and in chromosome 6 in mice. The gene encodes a type I membrane protein with a molecular weight of 70kDa [64]. This locus is adjacent and has a similar intron/exon organization to that of the CD4 co-receptor, suggesting that lag-3 and CD4 genes evolved via gene duplication from a pre-existing common ancestor gene. The mature protein consists of four extracellular immunoglobulin-like domains, a TM region containing a cleavage site mediated by two metalloproteases (ADAM 10 and ADAM 17) induced by TCR signaling, and a cytoplasmatic tail that mediates the intracellular transduction of inhibitory signals [64][65][66]. In addition, the LAG-3 cytoplasmic domain influences the protein membrane location. After TCR engagement LAG-3 co-localizes with CD3, CD4 or CD8 in the immunological synapses [67]. Apart from the cell membrane, LAG-3 is stored in lysosomes and can be translocated to the membrane after T-cell activation [68]. MHC-II molecules are considered canonical LAG-3 ligands and are in competition with CD4 binding. The engagement with its ligand reduces T-cell activity and cytokine secretion, blocking T-cell activation and function [18]. In addition, LAG-3 can bind Galectin-3 (Gal-3), a lectin expressed in various tumors and in activated T-cells. In tumors, Gal-3/LAG-3 binding inhibits anti-tumor-specific immune response by suppressing CD8 cytotoxic function and via the inhibition of plasmacytoid dendritic cell expansion [69]. Recently the fibrinogen-like protein1 (FGL1), a member of the fibrinogen family, has been identified as another LAG-3 ligand and has been proposed as an alternative mechanism of immune evasion. FGL1 expression is induced by IL-6, and it is highly upregulated in many solid tumors and is associated with poor prognosis and resistance to PD1 therapy [70]. The molecule is expressed on activated CD4+ and CD8+ T-cells, Tregs, NK cells, B-cells and dendritic cells [35]. On CD4+ cells, LAG-3 is present in Th0 and Th1 but not by Th2 [64]. Like other checkpoint molecules, LAG-3 has been identified in Tregs in the cancer microenvironment, in both natural and inducible activated subsets [71], where the IL-27/LAG-3 axis enhances the suppressive function [72]. The frequent co-expression with PD1 suggests a comparable function to PD1 [19][36]. In tumors, LAG-3 is expressed by exhausted CD8+/PD-1+ tumor-infiltrating lymphocytes [73], but its role in immune escape is still controversial. In B-cells, LAG-3 expression is induced by T-cells [74]. In addition, the molecule has been found in a subset of natural regulatory plasma cells (LAG-3/CD138 high) [75] and chronic lymphocytic leukemia [76].
In solid cancers, LAG-3 and PD-1 co-expression has been associated with poor sensitivity to PD-1 blockade and has been proposed as a biomarker for predicting the efficacy of immunotherapy, while data relating to AML are still scarce [34][77][78]. It should be remembered that the expression of MHC class II in AML blasts can be involved in both immune suppression and antigen presentation processes. Antibodies targeting LAG-3 are currently being tested in solid tumors, lymphomas and multiple myeloma, either in monotherapy or in combination with PD-1 inhibitors [18][35], with encouraging results, especially in metastatic melanoma, where the combination nivolumab + relatlimab significantly improved PFS. At present, only one clinical trial has been activated in AML patients: the AARON study (NCT04913922), which will test the safety and tolerability of a triplet of AZA, nivolumab (anti-PD-1) and relatlimab (anti-LAG-3) in patients with relapsed/refractory AML and newly diagnosed AML aged > 65 years: recruitment started in November 2022; no results are still available.
2.5. The CD200/CD200R Axis
CD200 is a highly conserved 48 kDa type-1 trans membrane glycoprotein, structurally related to the B7 family, encoded on chromosome 3q12-q13, which is close to the region coding for CD80/CD86 proteins. Through interaction with its receptor CD200R, CD200 causes the weakening of many immune-responsive effects, resulting in the prolonged survival of transplanted allograph, but also in tolerance toward tumor cells. CD200 overexpression has been reported both in solid tumors and in AML, where it marks LSCs but not the normal stem cell counterpart [35][79]. CD200 overexpression in AML has been associated with worse outcomes, mainly in the favorable prognostic groups [80][81]. CD200 expression on leukemic cells suppresses memory T-cell function, expands Tregs, and downmodulates NK function [82][83][84]. “In vitro” and “in vivo” murine models clearly demonstrated that the inhibition of the CD200/CD200R axis with MoAbs restores anti-AML immune response [85]. At present, the anti-CD200 MoAb samalizumab is being tested in solid tumors. In hematological malignancies, a phase I trial in CLL and multiple myeloma (NCT00648739) has been prematurely terminated for administrative reasons, not for safety concerns. At present, the only active trial of samalizumab in AML (the Beat AML trial, NCT03013998) has completed the recruitment.
2.6. The CD27/CD70 Axis
Among costimulatory pathways, CD27/CD70 has gained interest after demonstrating that it acts as a switch between immunity and tolerance. CD27, a member of the tumor necrosis factor superfamily, acts as a potent costimulatory molecule, providing a second signal for T-cell activation [86]. Different from other costimulatory molecules, it is constitutively expressed on resting naïve and memory T-cells and in a subset of NK cells [86], as well on memory B-cells and in most B-cell lymphomas [87]. CD27 signal is controlled by its unique ligand CD70, which is transiently upregulated on immune cells upon activation, but that is not expressed in normal tissues and hematopoietic system during homeostasis, suggesting that the early hematopoietic stages are independent of the CD27/CD70 axis [88]. There is evidence that CD27 engagement may produce different results depending on the strength, duration, and timing of the stimulation. After a short triggering, CD27/CD70 interaction provides a second signal for the differentiation of naïve CD4+ T-cells into Th1 effectors and of CD8+ T-cells into cytotoxic lymphocytes [89][90][91]. Conversely, the chronic or persistent triggering of CD27 leads to T-cell exhaustion and activation-induced cell death [92]. Moreover, chronic CD27/CD70 engagement maintains DCs in a tolerogenic state and favors the expansion of peripheral Tregs, suppressing anti-tumor immune responses [93]. Upon activation by CD70, the extracellular domain of CD27 is cleaved and is found in a soluble fragment (sCD27) in body fluids [94]. In tumors, an aberrant CD70 expression has been found without (solid tumors) or with (hematologic malignancies) CD27 co-expression, facilitating immune escape and contributing to generating a BM-suppressive microenvironment [94]. In LSCs, aberrant CD27 expression was detected in AML and in chronic myeloid leukemia (CML), and high levels of sCD27 were associated with a poor prognosis in terms of AML [95]. In LSCs, the CD27/CD70 pathway induces the aberrant activation of the Wnt pathway, which promotes LSC proliferation, drug resistance and disease progression [95][96]. Furthermore, the CD27/CD70 pathway can affect blast survival by regulating the MEK pathway, transcription factor AP-1 and the Wnt pathway through the activation of β-catenin [95]. With these premises, targeting the CD27/CD70 axis may constitute an effective immunotherapeutic strategy. However, the activity of anti-CD70 antibody–drug conjugates relies on the internalization of the drug, which can significantly differ among tumor cells [97]. More promising are antibodies that are able to induce cellular cytotoxicity (ADCC). The anti-CD79 antibody cusatuzumab (ARGX-110) showed the ability to elicit both ACC and ADCC because of afucosylation that enhances the binding to FcγRIIIa in a phase I trial in advanced malignancies [98]. Xenograft transplantation models and in vitro experiments proved cusatuzumab activity in eliminating LSCs by inducing differentiation and increasing apoptosis [99]. Based on the preclinical data, a phase I/II trial assessing the safety and efficacy of cusatuzumab as monotherapy and in combination with AZA in untreated AML patients unfit for intensive therapy has been designed (NCT0030612). The best hematological response was CR/CRi (8/2 patients), and the median time to response was 3.9 months, and a durable response was observed in six patients, with median PFS not being reached. At present, another trial evaluating cusatuzumab and AZA in newly diagnosed AML or high-risk MDSs unfit for intensive therapy has been completed (NCT042415499), but the results are not yet available. Lastly, two clinical studies for newly diagnosed AMLs unfit for intensive chemotherapy evaluating the combination of cusatuzumab + AZA (CULMINATE trial—NCT04023526) and cusatuzumab + AZA + venetoclax (ELEVATE trial—NCT04150887) are active but not recruiting.
References
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531.
- DiNardo, C.; Lachowiez, C. Acute Myeloid Leukemia: From Mutation Profiling to Treatment Decisions. Curr. Hematol. Malig. Rep. 2019, 14, 386–394.
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377.
- Stubbins, R.J.; Francis, A.; Kuchenbauer, F.; Sanford, D. Management of Acute Myeloid Leukemia: A Review for General Practitioners in Oncology. Curr. Oncol. 2022, 29, 6245–6259.
- Dessie, G.; Derbew Molla, M.; Shibabaw, T.; Ayelign, B. Role of Stem-Cell Transplantation in Leukemia Treatment. Stem. Cells Cloning 2020, 13, 67–77.
- Kreidieh, F.; Abou Dalle, I.; Moukalled, N.; El-Cheikh, J.; Brissot, E.; Mohty, M.; Bazarbachi, A. Relapse after allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia: An overview of prevention and treatment. Int. J. Hematol. 2022, 116, 330–340.
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22.
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982.
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39.
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738.
- Lyu, G.Y.; Yeh, Y.H.; Yeh, Y.C.; Wang, Y.C. Mutation load estimation model as a predictor of the response to cancer immunotherapy. NPJ Genom. Med. 2018, 3, 12.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. Immunopharmacol. 2020, 80, 106221.
- Chen, C.; Liang, C.; Wang, S.; Chio, C.L.; Zhang, Y.; Zeng, C.; Chen, S.; Wang, C.; Li, Y. Expression patterns of immune checkpoints in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 28.
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725.
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33.
- Kim, N.; Kim, H.S. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front. Immunol. 2018, 9, 2041.
- Boddu, P.; Kantarjian, H.; Garcia-Manero, G.; Allison, J.; Sharma, P.; Daver, N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk. Lymphoma 2018, 59, 790–802.
- Zhong, R.K.; Loken, M.; Lane, T.A.; Ball, E.D. CTLA-4 blockade by a human MAb enhances the capacity of AML-derived DC to induce T-cell responses against AML cells in an autologous culture system. Cytotherapy 2006, 8, 3–12.
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153.
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588.
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288.
- Orskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Gronbaek, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626.
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499.
- Gandini, S.; Massi, D.; Mandala, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98.
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481.
- Tan, J.; Chen, S.; Lu, Y.; Yao, D.; Xu, L.; Zhang, Y.; Yang, L.; Chen, J.; Lai, J.; Yu, Z.; et al. Higher PD-1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia. Chin. J. Cancer Res. 2017, 29, 463–470.
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510.
- Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Zhu, L.; Zeng, H.; Schell, T.D.; Zheng, H. PD-1(hi)TIM-3+ T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015, 5, e330.
- Giannopoulos, K. Targeting Immune Signaling Checkpoints in Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 236.
- Berthon, C.; Driss, V.; Liu, J.; Kuranda, K.; Leleu, X.; Jouy, N.; Hetuin, D.; Quesnel, B. In acute myeloid leukemia, B7-H1 (PD-L1) protection of blasts from cytotoxic T cells is induced by TLR ligands and interferon-gamma and can be reversed using MEK inhibitors. Cancer Immunol. Immunother. 2010, 59, 1839–1849.
- Chen, X.; Liu, S.; Wang, L.; Zhang, W.; Ji, Y.; Ma, X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol. Ther. 2008, 7, 622–627.
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune checkpoint molecules in acute myeloid leukaemia: Managing the double-edged sword. Br. J. Haematol. 2018, 181, 38–53.
- Davis, K.L.; Agarwal, A.M.; Verma, A.R. Checkpoint inhibition in pediatric hematologic malignancies. Pediatr. Hematol. Oncol. 2017, 34, 379–394.
- Kadia, T.M.; Cortes, J.E.; Ghorab, A.; Ravandi, F.; Jabbour, E.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; Konopleva, M.; Kantarjian, H.M. Nivolumab (NIVO) Maintenance (mainT) in High-Risk (HR) Acute Myeloid Leukemia (AML) Patients. J. Clin. Oncol. 2018, 34, 7014.
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Hendrickson, S.; Brandt, M.; Pierce, S.; Gordon, T.; et al. Phase IB/II study of nivolumab with azacytidine (AZA) in patients (pts) with relapsed AML. J. Clin. Oncol. 2017, 36, 7026.
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-Center Phase 2 Study of Pembroluzimab (Pembro) and Azacitidine (AZA) in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) and in Newly Diagnosed (≥65 Years) AML Patients. Blood 2019, 134, 832.
- Ravandi, F.; Daver, N.; Garcia-Manero, G.; Benton, C.B.; Thompson, P.A.; Bothakur, G.; Kadia, T.; Boddu, P.C.; Alvarado, Y.; Jabbour, E.J.; et al. Phase 2 study of combination of cytarabine, idarubicin, and nivolumab for initial therapy of patients with newly diagnosed acute myeloid leukemia. Blood 2017, 130, 815.
- Zeidner, J.F.; Vincent, B.G.; Esparza, S.; Ivanova, A.; Moore, D.; Foster, M.C.; Coombs, C.; Jamieson, K.; Van Devente, H.W.; Blanchard, L.; et al. Final Clinical Results of a Phase II Study of High Dose Cytarabine Followed By Pembrolizumab in Relapsed/Refractory AML. Blood 2019, 134, 831.
- Zeidan, A.M.; Cavenagh, J.; Voso, M.T.; Taussig, D.; Tormo, M.; Boss, I.; Copeland, W.B.; Gray, V.E.; Previtali, A.; O’Connor, T.; et al. Efficacy and Safety of Azacitidine (AZA) in Combination with the Anti-PD-L1 Durvalumab (durva) for the Front-Line Treatment of Older Patients (pts) with Acute Myeloid Leukemia (AML) Who Are Unfit for Intensive Chemotherapy (IC) and Pts with Higher-Risk Myelodysplastic Syndromes (HR-MDS): Results from a Large, International, Randomized Phase 2 Study. Blood 2019, 134, 829.
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530.
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760.
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428.
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143.
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792.
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194.
- Goncalves Silva, I.; Gibbs, B.F.; Bardelli, M.; Varani, L.; Sumbayev, V.V. Differential expression and biochemical activity of the immune receptor Tim-3 in healthy and malignant human myeloid cells. Oncotarget 2015, 6, 33823–33833.
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014.
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem. Cell 2010, 7, 708–717.
- Folgiero, V.; Cifaldi, L.; Li Pira, G.; Goffredo, B.M.; Vinti, L.; Locatelli, F. TIM-3/Gal-9 interaction induces IFNgamma-dependent IDO1 expression in acute myeloid leukemia blast cells. J. Hematol. Oncol. 2015, 8, 36.
- Goncalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 2017, 22, 44–57.
- Li, C.; Chen, X.; Yu, X.; Zhu, Y.; Ma, C.; Xia, R.; Ma, J.; Gu, C.; Ye, L.; Wu, D. Tim-3 is highly expressed in T cells in acute myeloid leukemia and associated with clinicopathological prognostic stratification. Int. J. Clin. Exp. Pathol. 2014, 7, 6880–6888.
- Zahran, A.M.; Mohammed Saleh, M.F.; Sayed, M.M.; Rayan, A.; Ali, A.M.; Hetta, H.F. Up-regulation of regulatory T cells, CD200 and TIM3 expression in cytogenetically normal acute myeloid leukemia. Cancer Biomark. 2018, 22, 587–595.
- Tan, J.; Yu, Z.; Huang, J.; Chen, Y.; Huang, S.; Yao, D.; Xu, L.; Lu, Y.; Chen, S.; Li, Y. Increased PD-1+Tim-3+ exhausted T cells in bone marrow may influence the clinical outcome of patients with AML. Biomark. Res. 2020, 8, 6.
- Tan, J.; Huang, S.; Huang, J.; Yu, Z.; Chen, Y.; Lu, Y.; Li, Y.; Chen, S. Increasing Tim-3+CD244+, Tim-3+CD57+, and Tim-3+PD-1+ T cells in patients with acute myeloid leukemia. Asia Pac. J. Clin. Oncol. 2020, 16, 137–141.
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/Tim-3 expression level is higher in AML patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175.
- Xu, L.; Xu, J.; Ma, S.; Li, X.; Zhu, M.; Chen, S.; Han, Y.; Tang, X.; Fu, Z.; Qiu, H.; et al. High Tim-3 expression on AML blasts could enhance chemotherapy sensitivity. Oncotarget 2017, 8, 102088–102096.
- Darwish, N.H.; Sudha, T.; Godugu, K.; Elbaz, O.; Abdelghaffar, H.A.; Hassan, E.E.; Mousa, S.A. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: Potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget 2016, 7, 57811–57820.
- Zeidan, A.M.; Komrokji, R.S.; Brunner, A.M. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev. Anticancer. Ther. 2021, 21, 523–534.
- Zeidan, A.; Esteve, J.; Giagounidis, A.; Kim, H.-J.; Miyazaki, Y.; Platzbecker, U.; Schuh, A.C.; Sekeres, M.A.; Westermann, J.; Xiao, Z.; et al. The STIMULUS Clinical Trial Program: Evaluating Combination Therapy with MBG453 in Patients with Higher-Risk Myelodysplastic Syndrome (HR-MDS) or Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 2, S188.
- Brunner, A.; Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N. Anti-TIM-3 Antibody MBG453 in Combination with Hypomethylating Agents (HMAs) in Patients with High- Risk Myelodysplastic Syndrome (HR- MDS) and Acute Myeloid Leukemia: A Phase 1 Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, S188–S189.
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dreano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749.
- Li, N.; Workman, C.J.; Martin, S.M.; Vignali, D.A. Biochemical analysis of the regulatory T cell protein lymphocyte activation gene-3 (LAG-3; CD223). J. Immunol. 2004, 173, 6806–6812.
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504.
- Iouzalen, N.; Andreae, S.; Hannier, S.; Triebel, F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur. J. Immunol. 2001, 31, 2885–2891.
- Bae, J.; Lee, S.J.; Park, C.G.; Lee, Y.S.; Chun, T. Trafficking of LAG-3 to the surface on activated T cells via its cytoplasmic domain and protein kinase C signaling. J. Immunol. 2014, 193, 3101–3112.
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423.
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312.
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513.
- Do, J.S.; Visperas, A.; Sanogo, Y.O.; Bechtel, J.J.; Dvorina, N.; Kim, S.; Jang, E.; Stohlman, S.A.; Shen, B.; Fairchild, R.L.; et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal. Immunol. 2016, 9, 137–145.
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 2006, 108, 2280–2289.
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088.
- Lino, A.C.; Dang, V.D.; Lampropoulou, V.; Welle, A.; Joedicke, J.; Pohar, J.; Simon, Q.; Thalmensi, J.; Baures, A.; Fluhler, V.; et al. LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 2018, 49, 120–133.e9.
- Shapiro, M.; Herishanu, Y.; Katz, B.Z.; Dezorella, N.; Sun, C.; Kay, S.; Polliack, A.; Avivi, I.; Wiestner, A.; Perry, C. Lymphocyte activation gene 3: A novel therapeutic target in chronic lymphocytic leukemia. Haematologica 2017, 102, 874–882.
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; De Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat. Commun. 2019, 10, 1065.
- Chen, Y.; Tan, J.; Huang, S.; Huang, X.; Huang, J.; Chen, J.; Yu, Z.; Lu, Y.; Weng, J.; Du, X.; et al. Higher frequency of the CTLA-4+ LAG-3+ T-cell subset in patients with newly diagnosed acute myeloid leukemia. Asia. Pac. J. Clin. Oncol. 2020, 16, e12–e18.
- Ho, J.M.; Dobson, S.M.; Voisin, V.; McLeod, J.; Kennedy, J.A.; Mitchell, A.; Jin, L.; Eppert, K.; Bader, G.; Minden, M.D.; et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020, 4, 5402–5413.
- Damiani, D.; Tiribelli, M.; Raspadori, D.; Sirianni, S.; Meneghel, A.; Cavalllin, M.; Michelutti, A.; Toffoletti, E.; Geromin, A.; Simeone, E.; et al. Clinical impact of CD200 expression in patients with acute myeloid leukemia and correlation with other molecular prognostic factors. Oncotarget 2015, 6, 30212–30221.
- Tonks, A.; Hills, R.; White, P.; Rosie, B.; Mills, K.I.; Burnett, A.K.; Darley, R.L. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia 2007, 21, 566–568.
- Coles, S.J.; Hills, R.K.; Wang, E.C.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia 2012, 26, 2146–2148.
- Coles, S.J.; Gilmour, M.N.; Reid, R.; Knapper, S.; Burnett, A.K.; Man, S.; Tonks, A.; Darley, R.L. The immunosuppressive ligands PD-L1 and CD200 are linked in AML T-cell immunosuppression: Identification of a new immunotherapeutic synapse. Leukemia 2015, 29, 1952–1954.
- Morgan, H.J.; Rees, E.; Lanfredini, S.; Powell, K.A.; Gore, J.; Gibbs, A.; Lovatt, C.; Davies, G.E.; Olivero, C.; Shorning, B.Y.; et al. CD200 ectodomain shedding into the tumor microenvironment leads to NK cell dysfunction and apoptosis. J. Clin. Investig. 2022, 132, e150750.
- Rastogi, N.; Baker, S.; Man, S.; Uger, R.A.; Wong, M.; Coles, S.J.; Hodges, M.; Gilkes, A.F.; Knapper, S.; Darley, R.L.; et al. Use of an anti-CD200-blocking antibody improves immune responses to AML in vitro and in vivo. Br. J. Haematol. 2021, 193, 155–159.
- Hintzen, R.Q.; Lens, S.M.; Lammers, K.; Kuiper, H.; Beckmann, M.P.; van Lier, R.A. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J. Immunol. 1995, 154, 2612–2623.
- van Oers, M.H.; Pals, S.T.; Evers, L.M.; van der Schoot, C.E.; Koopman, G.; Bonfrer, J.M.; Hintzen, R.Q.; von dem Borne, A.E.; van Lier, R.A. Expression and release of CD27 in human B-cell malignancies. Blood 1993, 82, 3430–3436.
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The cloning of CD70 and its identification as the ligand for CD27. J. Immunol. 1994, 152, 1756–1761.
- van Oosterwijk, M.F.; Juwana, H.; Arens, R.; Tesselaar, K.; van Oers, M.H.; Eldering, E.; van Lier, R.A. CD27-CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int. Immunol. 2007, 19, 713–718.
- Peperzak, V.; Veraar, E.A.; Keller, A.M.; Xiao, Y.; Borst, J. The Pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J. Immunol. 2010, 185, 6670–6678.
- Taraban, V.Y.; Rowley, T.F.; Kerr, J.P.; Willoughby, J.E.; Johnson, P.M.; Al-Shamkhani, A.; Buchan, S.L. CD27 costimulation contributes substantially to the expansion of functional memory CD8+ T cells after peptide immunization. Eur. J. Immunol. 2013, 43, 3314–3323.
- Tesselaar, K.; Arens, R.; van Schijndel, G.M.; Baars, P.A.; van der Valk, M.A.; Borst, J.; van Oers, M.H.; van Lier, R.A. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat. Immunol. 2003, 4, 49–54.
- Muth, S.; Klaric, A.; Radsak, M.; Schild, H.; Probst, H.C. CD27 expression on Treg cells limits immune responses against tumors. J. Mol. Med. 2022, 100, 439–449.
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10.
- Riether, C.; Schurch, C.M.; Buhrer, E.D.; Hinterbrandner, M.; Huguenin, A.L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J. Exp. Med. 2017, 214, 359–380.
- Riether, C.; Schurch, C.M.; Flury, C.; Hinterbrandner, M.; Druck, L.; Huguenin, A.L.; Baerlocher, G.M.; Radpour, R.; Ochsenbein, A.F. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci. Transl. Med. 2015, 7, 298ra119.
- Nejadmoghaddam, M.R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23.
- Aftimos, P.; Rolfo, C.; Rottey, S.; Offner, F.; Bron, D.; Maerevoet, M.; Soria, J.C.; Moshir, M.; Dreier, T.; Van Rompaey, L.; et al. Phase I Dose-Escalation Study of the Anti-CD70 Antibody ARGX-110 in Advanced Malignancies. Clin. Cancer Res. 2017, 23, 6411–6420.
- Riether, C.; Pabst, T.; Hopner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Muller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat. Med. 2020, 26, 1459–1467.
More
Information
Subjects:
Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
618
Revisions:
3 times
(View History)
Update Date:
30 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No