Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Daniela Damiani and Version 3 by Rita Xu.
The prognosis of acute myeloid leukemia (AML) remains unsatisfactory. Among the reasons for the poor response to therapy and high incidence of relapse, there is tumor cell immune escape, as AML blasts can negatively influence various components of the immune system, mostly weakening T-cells.
The prognosis of acute myeloid leukemia (AML) remains unsatisfactory. Among the reasons for the poor response to therapy and high incidence of relapse, there is tumor cell immune escape, as AML blasts can negatively influence various components of the immune system, mostly weakening T-cells.
- acute myeloid leukemia
- immune microenvironment
- checkpoint inhibitors
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease in which conventional treatment is still associated with unacceptably high rates of relapse and death despite complete remission (CR) being obtained in around 70% of patients [1]. The disease results from the transformation of rapidly proliferating progenitor cells. In AML, dysfunctional hierarchy prevails over the normal one, resulting in progressive cytopenias and increased risks of bleeding and infections.
In recent years, much progress has been made concerning the knowledge of the molecular pathogenesis of AML [2][3][2,3], and new drugs acting on various genetic abnormalities have been developed, thus permitting novel therapeutic approaches and, eventually, higher survival rates [4]. The treatment strategies used for AML depend on many factors, both disease-specific (e.g., prior exposure to toxics, prior myelodysplastic syndrome—MDS—cytogenetic and molecular abnormalities, and relapsed or refractory disease) and patient-specific (e.g., age, comorbidities, and previous exposure to chemotherapy). The current treatment for most cases of AML, excluding acute promyelocytic leukemia, consists of two phases: CR induction and the consolidation phase. In patients eligible for intensive induction, the “classical” 3 + 7 regimen is generally used; alternatives include the addition of fludarabine (FLAI) or mitoxantrone-based regimens [5]. In addition, the use of the FLT3 inhibitor midostaurin has become a standard in AML patients harboring FLT3-ITD mutation [5][6][5,6]. In patients not eligible for intensive chemotherapy, hypomethylating agents (HMAs), alone or in association with the BCL2 inhibitor venetoclax or IDH1 inhibitor ivosidenib, are the most recent options [5]. In “fit” patients, consolidation includes intermediate or high-dose cytarabine and allogeneic hematopoietic cell transplantation (HCT), which is still considered the only curative option [7]. However, the “cure” of AML remains a major challenge, as almost half of transplanted patients relapse after HCT, a risk that depends on disease status at transplant, donor type, T cell depletion strategies, and conditioning regimen [8]. It has now become evident that AML development and progression, as well as drug resistance acquisition, are associated with changes in the bone marrow microenvironment, which provide physical protection and release pro-survival factors for leukemic cells and induce dysregulated immune responses that facilitate the immune escape of AML blasts [9]. In addition to targeting molecular alterations, the manipulation of the AML immune microenvironment to restore immune surveillance appears to be an attractive strategy.
2. Checkpoint Inhibitors in AML Therapy
The evidence that immune checkpoint interactions contribute to AML immune evasion, not only at relapse but also in newly diagnosed disease, represents the rationale to use this therapeutic approach to reactivate immune sensitivity through a block of co-inhibitory ligands. From the approval of the first antibody blocking an immune checkpoint in 2011, immune checkpoint inhibitors (ICI) revolutionized the oncology field, resulting in durable response rates in patients with historically limited therapeutic options [10][107]. After approval in solid malignancies, various molecules, such as ipilimumab, pembrolizumab, cemiplimab, avelumab, and durvalumab, acting on specific targets, are undergoing clinical trials in relation to AML [11][108]. Due to their limited activity when used as a monotherapy, despite the demonstrated ability of co-inhibitory blockade and improved immune response, combination trials are ongoing. Despite the promising activity of checkpoint inhibitors in solid tumors [12][109], so far, clinical trials relating to AML have reported controversial results, possibly due to the low mutational burden and the DNA mismatch repair proficiency in AML when compared to solid tumors [13][14][110,111]. However, about fifty trials are currently ongoing testing ICI in AML and MDS, either as monotherapy or in combination with chemotherapy or HMAs. Key ICI potential targets in AML are shown in Figure 1.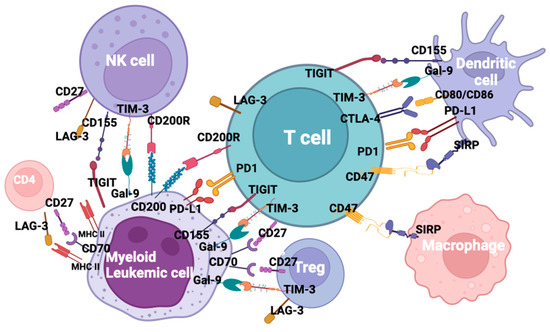
Figure 1. Checkpoint inhibitors and ligands in acute myeloid leukemia.
2.1. The CTLA-4/B7 Axis
CTLA-4 (CD152) is a member of the immunoglobulin-related receptors interacting with CD80 and CD86 ligands to deliver an inhibitory signal aimed at terminating immune responses. Moreover, it has a role in Tregs induction and, consequently, in regulating tolerance and autoimmunity [15][112]. The aberrant expression of CTLA-4 has been reported in AML, with a detrimental effect on disease outcomes [16][113]. In animal models, blocking CTLA-4 enhanced T-cell activity and suppressed Tregs [17][114]. In clinical trials relating to melanoma, anti-CTLA-4 ipilimumab increased the Teff/Tregs ratio, enhanced NK activity and restored T-effector function, ultimately prolonging survival [18][19][115,116]. Data on AML cell lines and preliminary results from clinical trials confirmed what has been reported in solid tumors [20][117]. Zhong et al. have shown that anti-CTLA-4 can improve AML-specific T-cell function in terms of frequency, cytotoxic capacity and IFN-γ secretion [21][118]. Data on the “in vivo” efficacy of ipilimumab are scarce. Davids and coll. reported rates of CR and partial response (PR) of 23% and 9%, respectively, in hematological malignancies receiving single-agent ipilimumab for relapse after HCT [22][119]. Bashey et al. observed no responses in AML patients among a small cohort of 29 patients with various hematologic malignancies relapsed after HCT [23][120]. A phase I trial of ipilimumab and a definite dose of Treg-depleted donor lymphocyte infusion (DLI) for AML and MDS relapsed after HCT is recruited (NCT03912064). HMAs have been shown to upregulate checkpoint receptors and their ligands in AML/MDS models [24][121]. The “in vitro” data correlates to a clinical study in AML, showing that the azacytidine (AZA)-induced demethylation of PD-1 promoter translates into the increased expression of PD-1 and an unfavorable outcome in the absence of immunotherapy [25][122]. A phase I trial combining ipilimumab and decitabine (DAC) in relapsed/refractory AML and MDS after allogeneic HCT or HCT-naïve is currently active but not recruiting (NCT2890329).2.2. The PD1/PD-L1 Axis
Programmed death receptor-1 (PD1 or CD279) is a type I transmembrane protein expressed mostly in activated immune cells [26][105]. PD1 binds two ligands: PD-L1 (CD274) and PD-L2 (CD273). PD-L1 is a member of the B7 family of co-stimulatory/co-inhibitory molecules present in hematopoietic cells and is upregulated or aberrantly expressed in many tumors [26][27][105,123]. In AML, the up-regulation of PD1 was reported in T-effector cells and in Tregs [28][106]; it is hypothesized that PD1 overexpression on CD8+ T-cells may account for CTL dysfunction and immune suppression during AML progression [29][124]. Moreover, PD1 and T-cell immunoglobulin and mucin domain 3 (TIM-3) co-expression on T-cells has been correlated to T-cell exhaustion in human and murine models [30][125] and can predict leukemia relapse post-HCT [31][126]. In addition, the co-expression of PD1 and TIM-3 seems to be more frequent in patients experiencing multiple relapses [28][106]. On the other hand, the up-regulation of PD-L1 and PD-L2 at diagnosis, relapse and during treatment correlates with resistance to therapy and poor prognosis [32][33][34][35][127,128,129,130]. PD1/PD-L1 engagement drives an inhibitory signal causing T-cell exhaustion and favoring the immune escape of neoplastic cells [26][105]. Moreover, it induces the apoptosis of tumor-specific cells and favors Tregs differentiation and resistance to CD8+ mediated cytolysis [36][131]. Three PD1-blocking antibodies (nivolumab, pembrolizumab and cemiplimab) and three PD-L1 inhibitors (atezolizumab, avelumab and durvalumab) have been approved to date by the FDA for various solid tumors [11][108]. However, PD1 and PD-L1 inhibitors have still not been approved for AML. The results of a few clinical trials of drugs against the PD1–PD-L1 axis that have been completed or are ongoing in relation to AML are summarized in Table 1. Nivolumab was used alone as maintenance in high-risk AML [37][132] or to eliminate MRD. An ongoing phase II trial is exploring the tolerability of nivolumab in relapsed AML or MRD-positive AML after HCT (NCT03146468), while a phase I/Ib was terminated due to early toxicity. Additionally, pembrolizumab has been evaluated as a maintenance therapy in elderly AML patients or as a salvage therapy after relapse post-allogeneic transplant. Many clinical trials combining epigenetic therapies with ICI are currently recruiting (NCT03969446, NCT02996474, NCT4277442) [38][133]. However, patients who received prior HMA therapy displayed lower ORR compared to HMA-naïve patients (22% vs. 58%), suggesting that the combination of HMA and ICI may have better efficacy when administered early in the course of the disease. Daver et al. presented the interim results of the NCT02397720 trial, in which patients with relapsed/refractory AML or elderly patients with newly diagnosed AML were parallel assigned to receive AZA + nivolumab or AZA + nivolumab + ipilimumab: in the latter cohort, ORR and CR/PR rates were 44% and 36%, respectively. The combination of AZA and pembrolizumab in relapsed/refractory AML and in newly diagnosed AML patients over 65 years was investigated in the phase II trial NCT02845297, showing modest clinical activity [39][134]. Another phase II trial of nivolumab, cytarabine and idarubicin in newly diagnosed AML and MDS (NCT02464657) reported that responders had no evidence of MRD at the time of response, and non-responders were more likely to have TP 53 mutations and a higher frequency of BM CD4+ cells co-expressing PD1 and TIM-3 [40][135]. The phase II NCT02768792 trial is evaluating the efficacy of the association of pembrolizumab and high-dose cytarabine (HiDAC) in relapsed/refractory AML. The preliminary results showed that pretreatment CD8+ TCR diversity was associated with response, as well as increased chemokine receptors in peripheral blood CD8+ cells and the increased expression of genes involved in p53, IFN-γ and Il-6 pathways [41][136]. No trial is currently ongoing to test cemiplimab in AMLs. The preliminary results of the combination of AZA and durvalumab in MDS and AML, which are unfit for intensive chemotherapy, showed an ORR of 61.9% in MDS and of 31% in AML and median OS 11.6 months in MDS and 13.0 months in AML (NCT02775903) [42][137]. Many phases I and II trials investigating the PD-L1 inhibitors avelumab or atezolizumab in association with HMAs (AZA, DAC or guadecitabine) are recruiting (NCT02953561, NCT03395973, NCT02935361, NCT02892318, NCT3154827), but no results are available at present.Table 1. Summary of results of a few clinical trials of drugs against PD1–PD-L1 axis completed or ongoing in AML.
| Trial Identifier | Drug | Disease | Response | Survival | Ref. |
|---|---|---|---|---|---|
| NCT02532231 | Nivolumab | HR AML | CR 79% at 6 months, 71% at 12 months |
OS 86% at 12 mo., 67% at 18 mo. |
[37][132] |
| NCT02397720 | Nivolumab + AZA | RR AML | ORR 58%, CR/CRi 21%, HI 7% | Median OS 9.2 mo. | [38][133] |
| AZA-naïve AML | ORR 22% | ||||
| NCT02464657 | Nivolumab + cytarabine + idarubicin | Newly diagnosed AML and MDS | CR/CRi 80% | Median OS 18.5 mo. | [40][135] |
| NCT02845297 | Pembrolizumab + AZA | RR AML | ORR 32%, CR/CRi 14% | Median OS 10.8 mo. | [39][134] |
| Newly diagnosed elderly AML | CR/CRi 47% | Median OS 13.1 mo. | |||
| NCT02768792 | Pembrolizumab + cytarabine | RR AML | ORR 46%, CR/CRi 38% | [41][136] | |
| NCT02775903 | Durvalumab + AZA | Unfit MDS and AML | ORR 61.9% in MDS ORR 31% in AML |
Median OS 11.6 mo. MDS Median OS 13 mo. AML |
[42][137] |