Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesca Cottini | -- | 1273 | 2023-08-25 16:06:11 | | | |
| 2 | Fanny Huang | Meta information modification | 1273 | 2023-08-28 07:22:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Galloway, J.; Petrilla, C.; Kudalkar, R.; Ismael, A.; Cottini, F. Mutations and Biomarkers of DNA Damage in Myeloma. Encyclopedia. Available online: https://encyclopedia.pub/entry/48491 (accessed on 09 August 2026).
Galloway J, Petrilla C, Kudalkar R, Ismael A, Cottini F. Mutations and Biomarkers of DNA Damage in Myeloma. Encyclopedia. Available at: https://encyclopedia.pub/entry/48491. Accessed August 09, 2026.
Galloway, Joshua, Cole Petrilla, Ruchi Kudalkar, Aya Ismael, Francesca Cottini. "Mutations and Biomarkers of DNA Damage in Myeloma" Encyclopedia, https://encyclopedia.pub/entry/48491 (accessed August 09, 2026).
Galloway, J., Petrilla, C., Kudalkar, R., Ismael, A., & Cottini, F. (2023, August 25). Mutations and Biomarkers of DNA Damage in Myeloma. In Encyclopedia. https://encyclopedia.pub/entry/48491
Galloway, Joshua, et al. "Mutations and Biomarkers of DNA Damage in Myeloma." Encyclopedia. Web. 25 August, 2023.
Copy Citation
Multiple myeloma (MM) is a plasma cell malignancy characterized by several genetic abnormalities, including chromosomal translocations, genomic deletions and gains, and point mutations. DNA damage response (DDR) and DNA repair mechanisms are altered in MM to allow for tumor development, progression, and resistance to therapies.
multiple myeloma
DNA damage response
DNA repair
1. Introduction
Environmental stressors or biologic components such as reactive oxidative species (ROS) are known to induce mutations and genomic abnormalities [1]. Cancer cells, due to a high replication rate, oncogene activation, or ROS production, have a greater mutational burden than normal tissues [2]. As increasing genomic abnormalities emerge, cancer cells activate the DNA damage response (DDR) to either correct otherwise deteriorating DNA sequences (DNA repair pathways) or to undergo senescence or apoptosis [3]. Indeed, with the introduction of beneficial mutational abnormalities come unwanted gene mutations which hinder proliferation or survival. As DNA repair pathways are usually hyperactive in cancer cells, new inhibitors targeting these mechanisms are under development to stop or stall the progression of cancer by reducing DNA repair and hence increasing cell death [4].
Multiple myeloma (MM) is a plasma cell malignancy originating in the bone marrow of afflicted individuals [5]. Over 35,000 individuals are diagnosed yearly with MM in the United States, with a median age of diagnosis of 68 years. MM is characterized by different clinical scenarios, including fatigue, lytic bone lesions, and kidney damage. MM is a disease with a multistep development pathway, beginning with healthy differentiated plasma cells. A preliminary step in MM is monoclonal gammopathy of undetermined significance (MGUS), where abnormal plasma cells start producing a specific monoclonal protein (Figure 1), followed by a phase called smoldering MM (SMM). Although not all MGUS develop into MM, all cases of MM arise from MGUS [6]. MM karyotypes are usually complex, with numerical (hyperdiplody or hypodiplody) and structural abnormalities, such as chromosomal translocations, gains, or deletions of genomic loci or wider areas [7]. Research into specific genomic abnormalities using fluorescence in situ hybridization (FISH) methods have identified a variety of genomic aberrations, such as chromosomal translocations (e.g., t(11;14), t(4;14), t(14;16)), chromosomal deletions (del(17p13) or del(13q) or chromosomal gains (1q21+). Some of them, including chromosomal translocation t(11;14) or deletion 13q, are considered primary genetic events, also occurring in MGUS; others are considered secondary genetic events associated with progression to overt MM, such as MYC (MYC proto-oncogene, bHLH transcription factor) rearrangements, abnormalities in the DDR/DNA damage pathway (e.g., ataxia-telangiectasia mutated-ATM deletion, tumor protein p53-TP53 mutations, del(17p)), or mutations in genes of the mitogen-activated protein kinase (MAPK) pathway (KRAS proto-oncogene, GTPase-KRAS, NRAS proto-oncogene, GTPase-NRAS, B-Raf proto-oncogene, serine/threonine kinase-BRAF) [8][9] (Figure 1). Despite several therapeutic options, MM remains incurable, with poor outcomes in patients with high-risk features [10]. Autologous stem cell transplants (ASCTs) using melphalan, an alkylating agent, are still widely used due to their progression-free survival benefits [11][12]. However, recent studies have shown that melphalan increases the general mutational burden in MM cells [13], reopening the debate about the balance between DNA damage-mediated apoptosis and the repair of damaged DNA.
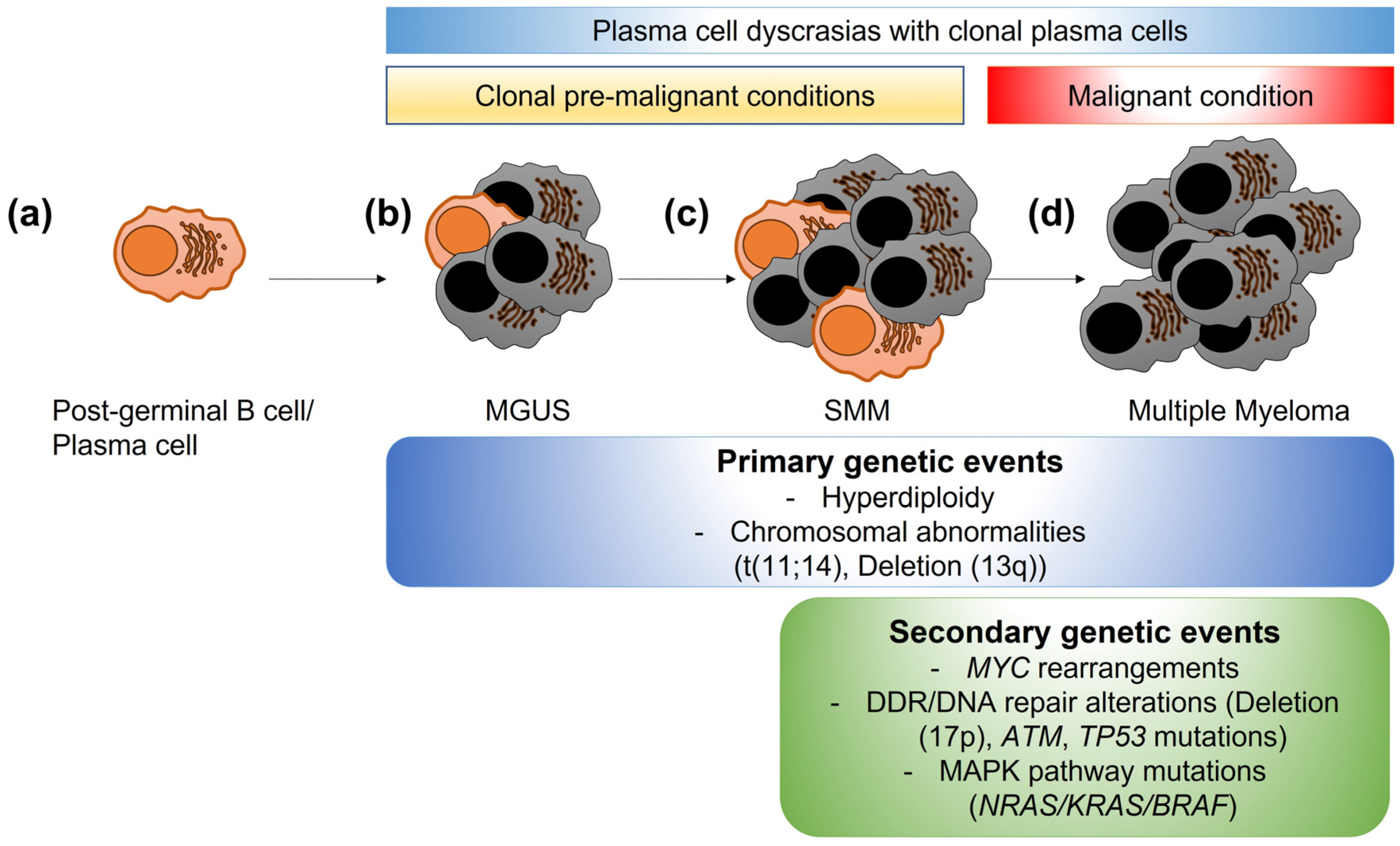
Figure 1. Clonal evolution of plasma cell dyscrasias. (a) A mature post-germinal B cell or plasma cell normally produces specific antibodies to aid in immune responses. (b) In some individuals, low levels of abnormal post-germinal B cells/plasma cells become clonally expanded (grey cells), producing a specific monoclonal-protein. This stage is called gammopathy of undetermined significance (MGUS) and is a premalignant condition. (c) Smoldering multiple myeloma (SMM) is the progression of MGUS, with higher numbers of abnormal clonal plasma cells. SMM is still a premalignant condition. (d) Multiple myeloma is the overt malignant condition, with higher percentages of abnormal clonal plasma cells leading to organ damage (anemia, kidney injury, bone lesions, or hypercalcemia). While some genetic changes are considered primary events also present in MGUS and SMM stages, secondary events, such as MYC rearrangements, alterations/mutations in DNA damage response or DNA repair genes, or mutations in the mitogen-activated protein kinase (MAPK) pathway genes are associated with progression to MM.
2. Mutations and Biomarkers of DNA Damage
2.1. Genomic Alterations Related to DDR and DNA Repair in MM
Loss-of-function mutations or deletion in ATM and in ATR have been reported in 2–4% of patients with sporadic MM [14][15], with cases of MM described in patients with ataxia telangiectasia. TP53 variants (either del(17p) or TP53 mutations) are present in 10–12% of cases at diagnosis and at higher rates at relapse, resulting in combined abnormalities in DDR in 15% of patients at diagnosis [15]. Patients with DDR abnormalities have reduced progression-free survival (PFS) and overall survival (OS), as described in the National Cancer Research Institute Myeloma XI trial [16]. Another specific mutational signature identified in MM which confers poor outcomes is the apolipoprotein B mRNA editing enzyme catalytic (APOBEC) signature [17][18]. The APOBEC3 family (A3A, A3B, A3C, and A3G) and activation-induced cytidine deaminase (AICDA/AID) are DNA-editing enzymes which act preferentially on single-strand DNA deaminating cytosines to uracil when cytosines immediately precede thymine (TpC context). This can lead to an enrichment of C > G and C > T mutations, a pattern associated with MAF bZIP transcription factor (MAF) translocations in MM [17][18]. Moreover, APOBEC3G via increasing HR activity is considered a driver for the acquisition of copy number variations and mutational changes in MM cells [19].
Polymorphism D693N within BRCA1 and polymorphisms in OGG1, ERCC1 (ERCC excision repair 1, endonuclease non-catalytic subunit), ERCC4 (ERCC excision repair 4, endonuclease catalytic subunit), XRCC1 (X-ray repair cross complementing 1), and XRCC2 (X-ray repair cross-complementing 2) genes, all enzymes involved in DNA repair, are associated with responses to high-dose melphalan [20]. Moreover, polymorphisms in genes of the BER pathway increased the risk of developing MM (e.g., OGG1 Ser326Cys) or conferred reduced OS (APEX1 Asp148Glu and mutY DNA glycosylase-MUTYH Gln324) in a series of Japanese patients with MM [21]. Finally, first-degree relatives of Ashkenazi Jewish carriers of common BRCA1 and BRCA2 mutations tend to develop MM more commonly than the general population [22], and at least one family with multiple cases of MM has been linked to BRCA2 mutations [23].
2.2. Biomarkers
High-dose melphalan is used as conditioning regimen for ASCT in myeloma. As with other alkylating agents, melphalan induces a range of cytotoxic and mutagenic adducts in DNA. Dimopoulos et al. confirmed the formation of monoadducts via melphalan therapy and reported that the area under the curve of total adducts in the peripheral blood mononuclear cells of patients post-melphalan is highly predictive of clinical responses [24]. However, melphalan also increases the general mutational burden in MM cells compared to bortezomib-lenalidomide-dexamethasone treatment [13][25]. Interestingly, patients achieving complete responses after ASCT have a significantly higher number of mutations than patients with less robust responses, possibly triggering neo-antigen formation and hence anti-tumoral immunity. However, the long-term repercussion of these mutations, especially in patients with previously unknown germline or acquired genetic abnormalities, is still vastly unexplored.
2.3. Relationship with Myeloid Neoplasm Conditions
Secondary primary malignancies (SPMs), especially acute leukemias or myelodysplastic syndrome, are not uncommon and occur with higher prevalence in patients with MM than in the general population [26]. This is partially due to host-related factors, genetic factors, and aging; however, an increase in the number of hematological SPMs has also been observed with co-exposure to lenalidomide and melphalan (especially with oral melphalan) [27]. Clonal hematopoiesis of indeterminate potential (CHIP), a condition characterized by the accumulation of leukemia-associated driver mutations in hematopoietic cells without underlying myeloid neoplasm [28], has also been reported in patients with MM at variable rates [29][30]. Interestingly, the presence of CHIP has been variably associated with MM outcomes or risk of development of therapy-related myeloid neoplasms, possibly due to the maintenance with lenalidomide post-ASCT as confounder [31][32]. Therefore, the relationship between genomic instability, lenalidomide/melphalan use, and onset of secondary myeloid conditions is complex and warrant further prospective studies.
References
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228.
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 2023, 23, 78–94.
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060.
- Kyle, R.A.; Rajkumar, S.V. Monoclonal gammopathy of undetermined significance. Br. J. Haematol. 2006, 134, 573–589.
- Robiou du Pont, S.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of Multiple Myeloma. J. Clin. Oncol. 2017, 35, 963–967.
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J. Clin. Oncol. 2020, 38, 2380–2389.
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013, 31, 4325–4332.
- Abdallah, N.; Rajkumar, S.V.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; et al. Cytogenetic abnormalities in multiple myeloma: Association with disease characteristics and treatment response. Blood Cancer J. 2020, 10, 82.
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320.
- Richardson, P.G.; Jacobus, S.J.; Weller, E.A.; Hassoun, H.; Lonial, S.; Raje, N.S.; Medvedova, E.; McCarthy, P.L.; Libby, E.N.; Voorhees, P.M.; et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N. Engl. J. Med. 2022, 387, 132–147.
- Samur, M.K.; Roncador, M.; Aktas Samur, A.; Fulciniti, M.; Bazarbachi, A.H.; Szalat, R.E.; Shammas, M.A.; Sperling, A.S.; Richardson, P.G.; Magrangeas, F.; et al. High-Dose Melphalan Treatment Significantly Increases Mutational Burden at Relapse in Multiple Myeloma. Blood 2023, 141, 1724–1736.
- Austen, B.; Barone, G.; Reiman, A.; Byrd, P.J.; Baker, C.; Starczynski, J.; Nobbs, M.C.; Murphy, R.P.; Enright, H.; Chaila, E.; et al. Pathogenic ATM mutations occur rarely in a subset of multiple myeloma patients. Br. J. Haematol. 2008, 142, 925–933.
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170.
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920.
- Maura, F.; Petljak, M.; Lionetti, M.; Cifola, I.; Liang, W.; Pinatel, E.; Alexandrov, L.B.; Fullam, A.; Martincorena, I.; Dawson, K.J.; et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2018, 32, 1044–1048.
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997.
- Talluri, S.; Samur, M.K.; Buon, L.; Kumar, S.; Potluri, L.B.; Shi, J.; Prabhala, R.H.; Shammas, M.A.; Munshi, N.C. Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma. Blood Cancer J. 2021, 11, 166.
- Dumontet, C.; Landi, S.; Reiman, T.; Perry, T.; Plesa, A.; Bellini, I.; Barale, R.; Pilarski, L.M.; Troncy, J.; Tavtigian, S.; et al. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transpl. 2010, 45, 1316–1324.
- Ushie, C.; Saitoh, T.; Iwasaki, A.; Moriyama, N.; Hattori, H.; Matsumoto, M.; Sawamura, M.; Isoda, J.; Handa, H.; Yokohama, A.; et al. The Polymorphisms of Base Excision Repair Genes Influence the Prognosis of Multiple Myeloma. Blood 2012, 120, 3981.
- Struewing, J.P.; Hartge, P.; Wacholder, S.; Baker, S.M.; Berlin, M.; McAdams, M.; Timmerman, M.M.; Brody, L.C.; Tucker, M.A. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N. Engl. J. Med. 1997, 336, 1401–1408.
- Lynch, H.T.; Ferrara, K.; Barlogie, B.; Coleman, E.A.; Lynch, J.F.; Weisenburger, D.; Sanger, W.; Watson, P.; Nipper, H.; Witt, V.; et al. Familial myeloma. N. Engl. J. Med. 2008, 359, 152–157.
- Dimopoulos, M.A.; Souliotis, V.L.; Anagnostopoulos, A.; Bamia, C.; Pouli, A.; Baltadakis, I.; Terpos, E.; Kyrtopoulos, S.A.; Sfikakis, P.P. Melphalan-induced DNA damage in vitro as a predictor for clinical outcome in multiple myeloma. Haematologica 2007, 92, 1505–1512.
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969.
- Mailankody, S.; Pfeiffer, R.M.; Kristinsson, S.Y.; Korde, N.; Bjorkholm, M.; Goldin, L.R.; Turesson, I.; Landgren, O. Risk of acute myeloid leukemia and myelodysplastic syndromes after multiple myeloma and its precursor disease (MGUS). Blood 2011, 118, 4086–4092.
- Palumbo, A.; Bringhen, S.; Kumar, S.K.; Lupparelli, G.; Usmani, S.; Waage, A.; Larocca, A.; van der Holt, B.; Musto, P.; Offidani, M.; et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: A meta-analysis of individual patient data. Lancet Oncol. 2014, 15, 333–342.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Chitre, S.; Stolzel, F.; Cuthill, K.; Streetly, M.; Graham, C.; Dill, C.; Mohamedali, A.; Smith, A.; Schetelig, J.; Altmann, H.; et al. Clonal hematopoiesis in patients with multiple myeloma undergoing autologous stem cell transplantation. Leukemia 2018, 32, 2020–2024.
- Wudhikarn, K.; Padrnos, L.; Lasho, T.; LaPlant, B.; Kumar, S.; Dispenzieri, A.; Lacy, M.; Rajkumar, S.V.; Gertz, M.; Mangaonkar, A.A.; et al. Clinical correlates and prognostic impact of clonal hematopoiesis in multiple myeloma patients receiving post-autologous stem cell transplantation lenalidomide maintenance therapy. Am. J. Hematol. 2021, 96, E157–E162.
- Tarek H. Mouhieddine; Adam S. Sperling; Robert Redd; Jihye Park; Matthew Leventhal; Christopher J. Gibson; Salomon Manier; Amin H. Nassar; Marzia Capelletti; Daisy Huynh; et al.Mark BustorosRomanos Sklavenitis-PistofidisSabrin TahriKalvis HornburgHenry DumkeMuhieddine M. ItaniCody J. BoehnerChia-Jen LiuSaud H. AlDubayanBrendan ReardonEliezer M. Van AllenJonathan J. KeatsChip StewartShaadi MehrDaniel AuclairRobert L. SchlossmanNikhil C. MunshiKenneth C. AndersonDavid P. SteensmaJacob P. LaubachPaul G. RichardsonJerome RitzBenjamin L. EbertRobert J. SoifferLorenzo TrippaGad GetzDonna S. NeubergIrene M. Ghobrial Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat. Commun. 2020, 11, 1-9.
- Lauren C. Peres; Christelle M. Colin-Leitzinger; Mingxiang Teng; Julie Dutil; Raghunandan Reddy Alugubelli; Gabriel DeAvila; Jamie K. Teer; Dongliang Du; Qianxing Mo; Erin M. Siegel; et al.Oliver A. HamptonMelissa AlsinaJason BrayerBrandon BlueRachid BazAriosto Siqueira SilvaTaiga NishihoriKenneth H. ShainNancy Gillis Racial and ethnic differences in clonal hematopoiesis, tumor markers, and outcomes of patients with multiple myeloma. Blood Adv. 2022, 6, 3767-3778.
More
Information
Subjects:
Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
730
Revisions:
2 times
(View History)
Update Date:
28 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No