Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Diego Schwarzstein | -- | 3135 | 2023-08-24 11:21:49 | | | |
| 2 | Lindsay Dong | Meta information modification | 3135 | 2023-08-25 02:46:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vargas-Uricoechea, H.; Nogueira, J.P.; Pinzón-Fernández, M.V.; Schwarzstein, D. Thyroid Antibodies in Autoimmune Thyroid Disease Diagnosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/48424 (accessed on 24 June 2026).
Vargas-Uricoechea H, Nogueira JP, Pinzón-Fernández MV, Schwarzstein D. Thyroid Antibodies in Autoimmune Thyroid Disease Diagnosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/48424. Accessed June 24, 2026.
Vargas-Uricoechea, Hernando, Juan Patricio Nogueira, María V. Pinzón-Fernández, Diego Schwarzstein. "Thyroid Antibodies in Autoimmune Thyroid Disease Diagnosis" Encyclopedia, https://encyclopedia.pub/entry/48424 (accessed June 24, 2026).
Vargas-Uricoechea, H., Nogueira, J.P., Pinzón-Fernández, M.V., & Schwarzstein, D. (2023, August 24). Thyroid Antibodies in Autoimmune Thyroid Disease Diagnosis. In Encyclopedia. https://encyclopedia.pub/entry/48424
Vargas-Uricoechea, Hernando, et al. "Thyroid Antibodies in Autoimmune Thyroid Disease Diagnosis." Encyclopedia. Web. 24 August, 2023.
Copy Citation
Autoimmune thyroid disease (AITD) refers to a spectrum of various diseases, with two extremes of clinical presentation, hypothyroidism (Hashimoto’s thyroiditis (HT) and hyperthyroidism (Graves–Basedow disease (GBD)). Both conditions are characterized by presenting a cellular and humoral autoimmune reaction, with an increase in the synthesis and secretion of antibodies directed toward various thyroid antigens, together with a phenomenon of thyrocyte necrosis and apoptosis (in HT) and a persistent thyrotropin-receptor stimulation (in GBD). The diagnosis of both entities is based on clinical, laboratory, and imaging findings.
thyroid
autoimmunity
antibodies
thyrotropin
receptor
1. Introduction
Immune tolerance is defined as a lack of response to an antigen, induced by previous exposure to said antigen; therefore, tolerance to self-antigens is a fundamental property of a “normal” immune system. Loss of tolerance to self-antigens leads to an inappropriate immune reaction called autoimmunity [1][2].
Diseases caused by such reactions are called autoimmune diseases (AIDs) and are characterized by pathogenic inflammatory responses induced by T lymphocytes (TL) and B lymphocytes (BL), which can induce “autotoxic” effects in virtually any organ or system [3][4].
When there is multi-organ involvement, the AID is classified as non-organ-specific (as in systemic lupus erythematosus and rheumatoid arthritis, among others), and when it affects a specific organ, it is classified as an organ-specific AID (such as type 1 diabetes, pernicious anemia, and autoimmune thyroid diseases (AITD), among others) [5][6].
Although the molecular mechanisms that induce AIDs are complex, it is clear that some genetic, non-genetic, epigenetic, and environmental factors are the basis for explaining the pathogenesis of these diseases and predicting clinical and biochemical responses [7][8].
AITD is the most common AID globally, presenting two classic phenotypes, hypothyroidism (subclinical or primary) in the context of Hashimoto’s thyroiditis (HT), or hyperthyroidism (subclinical or primary) in the context of Graves–Basedow disease (GBD) [9][10].
However, there are other thyroid conditions within the AITD group, such as postpartum thyroiditis, thyroiditis associated with autoimmune polyglandular syndromes, and drug-induced thyroiditis (for example, amiodarone), among others [11][12].
AITD is characterized by lymphocytic infiltration of the thyroid gland. For instance, in HT, the consequent inflammation induces follicular cell destruction, necrosis, and apoptosis, with subsequent fibrosis (and potentially hypothyroidism), with a humoral antibody (Ab)-mediated response directed against one or several thyroid antigens. These can include thyroid peroxidase (TPO) and thyroglobulin (Tg), among others [13][14].
In GBD, on the other hand, a humoral response predominates, with the presence of Abs that stimulate the thyrotropin (TSH) receptor (TSHR). It can be accompanied by goiter, hyperthyroidism, ophthalmopathy, and dermopathy (Figure 1) [15][16].
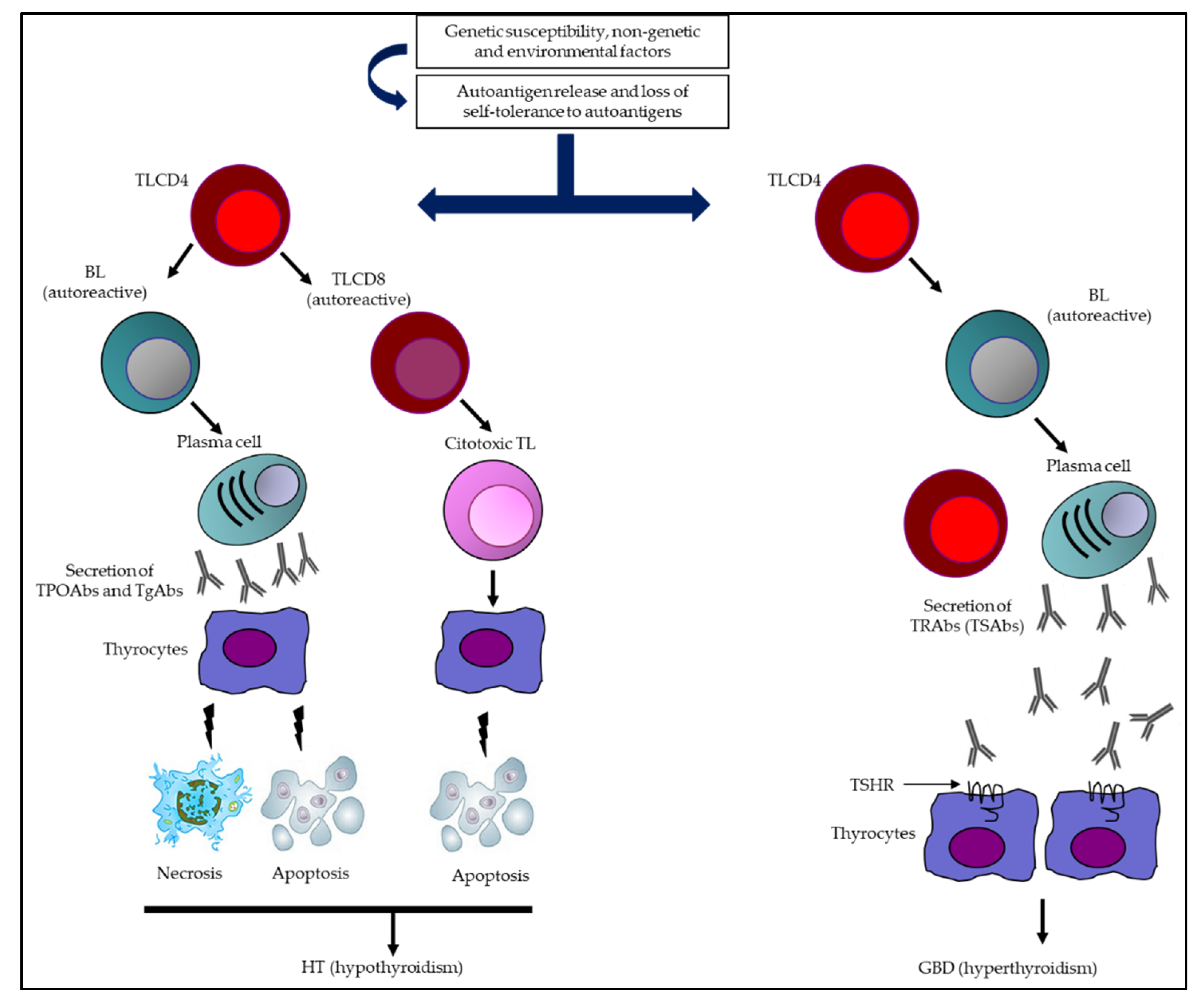
Figure 1. Summary of the immunological mechanisms of AITD leading to HT and GBD. Multiple genetic, epigenetic, non-genetic, and environmental factors come together in AITD, which (together with a loss of immune tolerance) are capable of inducing an immune response (humoral and cellular). In HT, the synthesis and secretion of TPOAbs and TgAbs, together with the activation of autoreactive TL, are capable of triggering destruction of thyrocytes (necrosis/apoptosis) and potentially hypothyroidism. Otherwise, in GBD, an extended humoral response predominates, with a greater capacity to secrete TRAbs (specifically TSAbs), which are the determinants of TSHR stimulation, inducing greater secretion of TH and, consequently, hyperthyroidism. Abbreviations: BL: B lymphocytes; GBD: Graves–Basedow disease; HT: Hashimoto’s thyroiditis; TgAbs: thyroglobulin antibodies; TH: thyroid hormones; TL: T lymphocytes (TL); TPOAbs: thyroid peroxidase antibodies; TRAbs: thyrotropin receptor antibodies; TSAbs: thyrotropin receptor-stimulating antibodies; TSHR: thyrotropin receptor.
2. Major Thyroid Antigens
2.1. Tg
The gene encoding Tg synthesis is a single copy gene (270 kb in length) located on chromosome 8q24.2–8q24.3, which contains an 8.5 kb coding sequence divided into 48 exons [17].
Tg is a glycosylated protein synthesized exclusively in the thyroid and is the largest (660 kDa) and most abundant autoantigen of the thyroid. It is essential for the synthesis of both thyroid hormones (TH) T4 and T3 since the synthesis of these hormones depends on the conformation, iodination, and post-translational modification of Tg [18].
TH synthesis (from Tg) occurs in the thyroid via iodination and coupling of pairs of tyrosines, which is completed by Tg proteolysis. Additionally, follicular Tg is capable of suppressing the thyroid feedback phenomenon since it can inhibit the expression of TTF-1, TTF-2, and PAX-8, decreasing the expression of the genes that code for the synthesis of TPO, NIS, TSHR, and Tg [18][19].
This suggests that Tg is not only the substrate for the synthesis of TH but also a regulator of thyroid function [18][19].
2.2. TPO
The gene encoding TPO synthesis is a single copy gene (2pter—p12) that codes for a protein of 933 amino acids, spanning the cell membrane, with a large extracellular domain (with 848 amino acids and five potential glycosylation sites) facing the follicular lumen and a cytoplasmic tail with a length of 61 amino acids [20][21].
The extracellular domain consists of three regions, which denote a high degree of sequence similarity, with other domains of specific three-dimensional structure, such as the myeloperoxidase-like domain, the complement control protein-like domain, and the epidermal growth factor-like domain. TPO expression is controlled by transcription factors, such as TTF-1, TTF-2, and PAX-8 [22][23].
TPO has two active sites, which facilitate the iodination of tyrosine residues in Tg, together with the dual oxidase enzyme and hydrogen peroxide, which subsequently allows for intrachain coupling of two iodotyrosine residues for TH synthesis. In addition, TPO catalyzes two reactions within the thyroid, the oxidation of iodine and the coupling of iodinated tyrosines in the process of TH synthesis [23][24].
Therefore, TPO plays a key role in the biosynthesis of TH and is a fundamental component of normal thyroid function.
2.3. TSHR
TSHR is encoded by a gene located at chromosome 14q31 and belongs to the family of G-protein-coupled receptors. It has a large extracellular domain (containing an N-terminal domain, a leucine-rich repeat domain, and a hinge region or cleavage domain), seven transmembrane passageways, and a small intracellular domain [25][26]).
TSHR couples to four G protein subfamilies, including Gs (inducing adenylyl cyclase activity and cAMP production), phospholipase C (which activates Gq/G11), G13 (which, in turn, is capable of inducing p44/42 mitogen-activated protein kinase), and Gi (which inhibits adenylyl cyclase activity) [27][28].
Mature TSHR contains two subunits (A and B). The A subunit is made up of a large extracellular domain mainly determined by multiple leucine-rich repeats and an N-terminal tail, where specific amino acids fold to form a complex TSH-binding pocket. The B subunit contains a short portion anchored to the membrane and the intracellular portion of the receptor [29][30].
TSHR expression occurs mainly on the basolateral membrane of thyrocytes, and its activation stimulates iodine uptake, TH synthesis and secretion, and thyrocyte proliferation [30][31].
3. Minor Thyroid Antigens
3.1. NIS
The gene that codes for the synthesis of NIS is located on chromosome 19p12–13.2 and encodes a glycoprotein of 643 amino acids with a molecular mass of about 70–90 kDa [32].
NIS is an intrinsic membrane protein belonging to the superfamily of sodium/solute symporters and the human transporter family (SLC5), containing 13 transmembrane domains, an extracellular amino terminus, and an intracellular carboxy terminus [32][33].
In thyrocytes, NIS is located at the basal cell level and is a mediator of active iodine transport to thyroid follicular cells involved in the first step of TH synthesis [34].
Iodine transport, mediated by NIS, is a vector process stimulated by TSH; NIS is also expressed in other tissues, such as the salivary glands, ductal cells, placenta, testicular cells, stomach, and mammary glands (during lactation). However, our understanding of the physiological role of the symporter in these tissues is not yet conclusive [34][35].
3.2. PDN
The gene encoding PDN synthesis is the same gene responsible for Pendred syndrome, which is an autosomal recessive disease that manifests with goiter and congenital sensorineural deafness. PDN is encoded by the SLC26A4 gene, which is located on chromosome 7q21-31 and contains 21 exons with an open reading frame of 2343 bp [36][37].
PDN (SLC26A4) is a glycoprotein composed of 780 amino acids. It contains three putative extracellular asparagine glycosylation sites and is considered an apical membrane-bound iodide transporter that acts as a multifunctional exchanger of multiple monovalent anions (for example, iodide, chloride, and bicarbonate) and is highly expressed both in the thyroid and in extrathyroid tissues (inner ear and kidneys). In the thyroid, PDN is expressed in the apical membrane of thyrocytes and participates in the transport of iodide to the colloid, indicating its importance in the TH synthesis [38][39][40].
3.3. Meg
Meg is a giant 600 kDa cell surface protein. Its gene covers 235,000 base pairs on the human chromosome 2q24-q31 and consists of 79 exons. It belongs to the endocytic low-density lipoprotein receptor family, which is expressed on the apical surface of thyrocytes; this expression is mediated by TSH. Meg has a high binding affinity for Tg and allows (at least in part) its uptake by thyrocytes; once Tg is internalized by Meg, lysosomal metabolism is avoided, and it is able to reach the basolateral membrane of the thyrocytes (via transcytosis), from where it is released into the blood [41][42][43].
4. Major Thyroid Abs
4.1. TgAbs
The ability of Tg to induce an immune response depends, at least in part, on the content of both T4 and T3. The concentration of TH within Tg is capable of changing its conformation, stimulating the formation of masked and unmasked epitopes. Consequently, the binding capacity of the Abs can be affected by the content of T4 and T3 in Tg [44].
TgAbs identified in individuals without AITD generally recognize highly conserved epitopes (located in the T4 and T3-containing regions of Tg), whereas, in individuals with AITD, TgAbs are less restricted. Additionally, TgAbs mainly recognize “conformational” and, to a lesser extent, “linear” epitopes, suggesting that the immunogenic potential of Tg increases to the extent that its fragments have a greater capacity to generate conformational epitopes [45][46].
4.2. TPOAbs
TPOAbs are considered the hallmark of AITD. The prevalence of positivity in individuals with AITD is higher than for TgAbs [47].
TPOAbs are capable of recognizing discontinuous determinants in TPO, which have been named immunodominant regions A (IDR-A) and B (IDR-B), and several contact residues constituting IDR-A and IDR-B have been identified: 225; 353–363; 377–386; 597–604; 611–618; 620; 624; 627; 630; 646; 707; 713–720; and 766–775 [48][49].
TPOAbs can also react against conformational or linear epitopes, and the polyclonal Abs present both in healthy individuals and those with AITD are directed against the same epitopes, taking into account that TPOAbs from healthy individuals do not block TPO action, while those identified in AITD patients can fix complement, produce lysis of thyrocytes, and competitively inhibit enzymatic activity [50].
4.3. TRAbs
Similar to TPOAbs being considered the hallmark of HT, TRAbs are the hallmark of GBD. The prevalence of TRAbs in subjects with HT is 10–20%, and in GBD, it is 90–95%; hence, its detection is recommended in the differential diagnosis of patients with hyperthyroidism [51].
The mechanism by which hyperthyroidism occurs in GBD is due to the presence of TRAbs, which simulate the effects of TSH on thyrocytes. TSHR is a receptor that belongs to the 7TM G-protein-coupled receptor family and is expressed in thyroid follicular cells (and also in thymocytes and retroorbital tissue fibroblasts) [52][53].
From the functional and biological points of view, TRAbs can be classified in three ways, stimulators, blockers, and neutral; for GBD, the most frequent are the stimulators. Stimulator TRAbs bind to the N-terminus of the TSH extracellular domain and consequently stimulate TH production (independently of the feedback phenomenon of the hypothalamic–pituitary–thyroid axis) [54][55].
TRAbs have a high receptor affinity; however, their absolute concentration is low. One explanation for this may be that they are produced by a limited number of BLs and APCs. Moreover, in some individuals, the immune response may alternate and change from a state, in which stimulatory TRAbs are initially (and predominantly) produced, to an opposite state, in which the production of blocking or neutral TRAbs is increased, resulting in changes in the clinical and biochemical findings of the disease [56][57].
TRAbs are a combination of highly related IgGs, which have the ability to bind to specific epitopes of the TSHR. However, these Abs can vary and fluctuate within the same individual (and between individuals). Therefore, the presence of subtle changes in the affinity or specificity of TRAbs can cause radical changes in their ability to activate the TSHR [57][58].
4.4. NISAbs and PDNAbs
The prevalence of positivity for NISAbs and PDNAbs in individuals with AITD and in the general population is highly variable. In general terms, the prevalence of NISAb positivity in healthy individuals is very low; however, its prevalence is increased in those with AITD (especially in GBD subjects) [59].
Moreover, some studies have documented that the prevalence of positivity for NISAbs and PDNAbs is similar in patients with AITD (prevalence close to 10% for each Ab). Likewise, the prevalence of PDNAbs positivity is only slightly higher in individuals with AITD (relative to healthy controls, being also detectable in the latter), and the prevalence is higher in individuals with GBD (versus HT and participants without AITD) [59][60].
Several explanations for these findings can be given, such as the type of population studied (populations with low prevalence of the disease), studies with small sample sizes, and the type of technology used to measure Abs. For these reasons, the role of NISAbs and PDNAbs in the diagnosis, prediction, and response and relapse rate of AITD still needs to be clarified.
4.5. MegAbs
As previously noted, Meg transports Tg through the thyroid epithelial cells, subsequently entering circulation in a Tg–Meg complex; thus, Meg is capable of eliciting an Ab-mediated immune response [61].
In rodent models of Heymann’s nephritis, Meg has the ability to induce Ab production and secretion (Heymann’s nephritis is an experimental rat model for active and passive immune-mediated nephritis). However, Meg, which is the target antigen, is localized in podocytes in the rat model, but in humans, megalin is found in the proximal tubule and not in podocytes [62][63].
This experimental model in rodents allowed us to propose that MegAbs could be generated and manifest in individuals with AITD in the same way. Initially, studies measuring IgG binding to L2 cells (a rat yolk sac carcinoma cell line known to express Meg) found a prevalence of 50% in subjects with HT and a lower percentage in individuals with GBD (10.5%), while it was not present in healthy individuals [64].
5. Clinical Utility of Thyroid Abs in AITD
5.1. Clinical Utility of TgAbs
Routine measurement of TgAbs in iodine-“sufficient” areas does not seem to be very useful as a screening method in the study of AITD; however, it may be useful in iodine-“deficient” areas, particularly in individuals with nodular goiter [65].
It has also been found that, in geographic regions where universal salt iodization programs (for consumption) have been developed, there has been a significant increase in the positivity of TgAbs after salt iodization, indicating that an eventual excess consumption of iodine (through salt) may also increase the immunogenicity of Tg, leading to a higher rate of thyroid autoimmunity. Therefore, it could be argued that in areas where programs of salt iodization have been implemented and where excess consumption has been documented, the measurement of TgAbs could play an important role in the population characterization of thyroid autoimmunity and the potential risk of developing thyroid functional disorders [9][66][67].
5.2. Clinical Utility of TPOAbs
In patients with hypothyroidism (subclinical or primary), TPOAb positivity determines the diagnosis of HT, while in euthyroid individuals, TPOAb positivity is associated with a significant increase in the risk of developing hypothyroidism over time [68][69].
Likewise, TPOAbs have been associated with the development of ocular alterations in patients with GBD. However, the results have been contradictory, especially in children. These findings have not been corroborated on a large scale in adults [14][70].
Moreover, TPOAbs have been related to a higher rate of infertility, premature birth, and spontaneous abortions (independent of TSH or T4 levels). In fact, it is recommended to evaluate TPOAbs levels in the pre-pregnancy period and once a pregnancy is confirmed since their presence (even with TSH values in the normal range of 2.5–4.0 mIU/L) could indicate the need for levothyroxine replacement [71].
5.3. Clinical Utility of TRAbs
The clinical utility of TRAbs in the diagnosis and monitoring of AITD remains controversial, despite the fact that their presence defines the diagnosis of GBD, especially in those individuals with long-standing hyperthyroidism associated with extrathyroid manifestations (ophthalmopathy, myxedema) [72].
Although in the diagnostic approach of hyperthyroidism (subclinical or primary), imaging methods such as ultrasound and/or thyroid scintigraphy can be used, the measurement of TRAbs can resolve the diagnosis in the majority of affected individuals, differentiating GBD from a “Hashitoxicosis” or other types of thyroiditis that debut with hyperthyroidism, or also fictitious thyrotoxicosis, and even toxic nodular goiter [73].
On the other hand, TRAbs can be detected in practically all patients with thyroid ophthalmopathy. In fact, TRAbs levels correlate with the severity and clinical activity of the disease, and in addition, a high level of TRAbs in patients with early ophthalmopathy predicts a poor prognosis. TRAbs are also useful in those individuals with clinical features of thyroid ophthalmopathy but with normal or discordant thyroid function (for example, with hypothyroidism) [74][75].
5.4. Clinical Utility of PDNAbs, NISAbs, MegAbs, and Other Abs
The utility of these Abs in the diagnosis, treatment, and prognosis of AITD remains to be demonstrated; to date, none have been shown to have superior diagnostic performance to that provided by TPOAbs, TgAbs, and TRAbs in individuals with HT and GBD [42][43][59][60][63][76].
Other Abs that have been described in patients with AITD are THAbs. THAbs are Abs that can bind to TH, and the presence of such Abs seems to be related to a massive “leakage” of Tg, exposing the immune system to several hormonogenic epitopes of the Tg and inducing a humoral (Ig-mediated) response. Four types of THAb have been described based on the presence of IgM or IgG (T4-IgM, T4-IgG, T3-IgM, and T3-IgG), with T4-IgG and T3-IgG being the most prevalent [77][78].
The occurrence of THAbs is variable; for example, in the general population, the prevalence is approximately 1%, but in individuals with AITD, the prevalence is 20–23% and 32–46% (in HT and GBD, respectively) [79][80].
6. Conclusions
TRAbs help in the diagnostic confirmation of GBD but also in differential diagnosis with other types of hyperthyroidism; additionally, they serve as a prognostic factor in thyroid ophthalmopathy, in the risk of relapse, and in the risk of fetal/neonatal hyperthyroidism, among other functions.
Despite the usefulness that thyroid Abs provide in the study of AITD, it must be taken into account that this condition is a clinical syndrome with multiple manifestations that may overlap; therefore, the integration of clinical, imaging, and laboratory components are those that will allow a diagnosis to be made with greater certainty. The usefulness of TRAbs in the clinical approach to GBD has been demonstrated; however, in HT, the mere presence of TPOAbs and/or TgAbs are not enough to establish the diagnosis since HT is defined as a histopathological diagnosis, and although TPOAbs and/or TgAbs correlate well with histopathologic findings, their mere presence is not always a disease condition.
Other thyroid Abs (such as NISAbs, PDNAbs, and MegAbs) have not yet been shown to have a higher diagnostic performance than major thyroid antibodies; therefore, their usefulness in studying AITD has yet to be demonstrated.
References
- Skevaki, C.; Wesemann, D.R. Antibody repertoire and autoimmunity. J. Allergy Clin. Immunol. 2023, 151, 898–900.
- Huffaker, M.F.; Sanda, S.; Chandran, S.; Chung, S.A.; Clair, E.W.S.; Nepom, G.T.; Smilek, D.E. Approaches to Establishing Tolerance in Immune Mediated Diseases. Front. Immunol. 2021, 12, 744804.
- Petersone, L.; Edner, N.M.; Ovcinnikovs, V.; Heuts, F.; Ross, E.M.; Ntavli, E.; Wang, C.J.; Walker, L.S.K. T Cell/B Cell Collaboration and Autoimmunity: An Intimate Relationship. Front. Immunol. 2018, 9, 1941.
- Corneth, O.B.J.; Neys, S.F.H.; Hendriks, R.W. Aberrant B Cell Signaling in Autoimmune Diseases. Cells 2022, 11, 3391.
- Samuels, H.; Malov, M.; Detroja, T.S.; Ben Zaken, K.; Bloch, N.; Gal-Tanamy, M.; Avni, O.; Polis, B.; Samson, A.O. Autoimmune Disease Classification Based on PubMed Text Mining. J. Clin. Med. 2022, 11, 4345.
- Hundt, J.E.; Hoffmann, M.H.; Amber, K.T.; Ludwig, R.J. Editorial: Autoimmune pre-disease. Front. Immunol. 2023, 14, 1159396.
- Chi, X.; Huang, M.; Tu, H.; Zhang, B.; Lin, X.; Xu, H.; Dong, C.; Hu, X. Innate and adaptive immune abnormalities underlying autoimmune diseases: The genetic connections. Sci. China Life Sci. 2023, 66, 1482–1517.
- Mazzone, R.; Zwergel, C.; Artico, M.; Taurone, S.; Ralli, M.; Greco, A.; Mai, A. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 2019, 11, 34.
- Bogusławska, J.; Godlewska, M.; Gajda, E.; Piekiełko-Witkowska, A. Cellular and molecular basis of thyroid autoimmunity. Eur. Thyroid. J. 2022, 11, e210024.
- McLachlan, S.M.; Rapoport, B. Discoveries in Thyroid Autoimmunity in the Past Century. Thyroid 2023, 33, 278–286.
- Rahimova, R.R. Autoimmune thyroiditis (review of literature). Russ. Clin. Lab. Diagn. 2022, 67, 286–291.
- Stasiak, M.; Lewiński, A. New aspects in the pathogenesis and management of subacute thyroiditis. Rev. Endocr. Metab. Disord. 2021, 22, 1027–1039.
- Vargas-Uricoechea, H. Molecular Mechanisms in Autoimmune Thyroid Disease. Cells 2023, 12, 918.
- Daramjav, N.; Takagi, J.; Iwayama, H.; Uchino, K.; Inukai, D.; Otake, K.; Ogawa, T.; Takami, A. Autoimmune Thyroiditis Shifting from Hashimoto’s Thyroiditis to Graves’ Disease. Medicina 2023, 59, 757.
- Wémeau, J.-L.; Klein, M.; Sadoul, J.-L.; Briet, C.; Vélayoudom-Céphise, F.-L. Graves’ disease: Introduction, epidemiology, endogenous and environmental pathogenic factors. Ann. Endocrinol. 2018, 79, 599–607.
- Chang, Y. Graves’ Disease is a Thyroid Autoimmune Disorder Identified by Excessive Thyroid Hormone Production. Thyroid. Disorders Ther. 2023, 12, 294.
- Coscia, F.; Taler-Verčič, A.; Chang, V.T.; Sinn, L.; O’reilly, F.J.; Izoré, T.; Renko, M.; Berger, I.; Rappsilber, J.; Turk, D.; et al. The structure of human thyroglobulin. Nature 2020, 578, 627–630.
- Citterio, C.E.; Rivolta, C.M.; Targovnik, H.M. Structure and genetic variants of thyroglobulin: Pathophysiological implications. Mol. Cell. Endocrinol. 2021, 528, 111227.
- Tosatto, L.; Coscia, F. A glance at post-translational modifications of human thyroglobulin: Potential impact on function and pathogenesis. Eur. Thyroid. J. 2022, 11, e220046.
- Godlewska, M.; Gawel, D.; Buckle, A.M.; Banga, J.P. Thyroid Peroxidase Revisited—What’s New? Horm. Metab. Res. 2019, 51, 765–769.
- Ruf, J.; Carayon, P. Structural and functional aspects of thyroid peroxidase. Arch. Biochem. Biophys. 2006, 445, 269–277.
- Godlewska, M.; Banga, P.J. Thyroid peroxidase as a dual active site enzyme: Focus on biosynthesis, hormonogenesis and thyroid disorders of autoimmunity and cancer. Biochimie 2019, 160, 34–45.
- Williams, D.E.; Le, S.N.; Godlewska, M.; Hoke, D.E.; Buckle, A.M. Thyroid Peroxidase as an Autoantigen in Hashimoto’s Disease: Structure, Function, and Antigenicity. Horm. Metab. Res. 2018, 50, 908–921.
- Mondal, S.; Raja, K.; Schweizer, U.; Mugesh, G. Chemistry and Biology in the Biosynthesis and Action of Thyroid Hormones. Angew. Chem. Int. Ed. 2016, 55, 7606–7630.
- Chu, Y.-D.; Yeh, C.-T. The Molecular Function and Clinical Role of Thyroid Stimulating Hormone Receptor in Cancer Cells. Cells 2020, 9, 1730.
- Vieira, I.H.; Rodrigues, D.; Paiva, I. The Mysterious Universe of the TSH Receptor. Front. Endocrinol. 2022, 13, 944715.
- Marín-Sánchez, A.; Álvarez-Sierra, D.; González, O.; Lucas-Martin, A.; Sellés-Sánchez, A.; Rudilla, F.; Enrich, E.; Colobran, R.; Pujol-Borrell, R. Regulation of TSHR Expression in the Thyroid and Thymus May Contribute to TSHR Tolerance Failure in Graves’ Disease Patients via Two Distinct Mechanisms. Front. Immunol. 2019, 10, 1695.
- Kleinau, G.; Vassart, G. TSH Receptor Mutations and Diseases. In Endotext ; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000.
- Gershengorn, M.C.; Osman, R. Molecular and cellular biology of thyrotropin-releasing hormone receptors. Physiol. Rev. 1996, 76, 175–191.
- Rapoport, B.; McLachlan, S.M. TSH Receptor Cleavage Into Subunits and Shedding of the A-Subunit; A Molecular and Clinical Perspective. Endocr. Rev. 2016, 37, 114–134.
- Helfinger, L.; Tate, C.G. Expression and Purification of the Human Thyroid-Stimulating Hormone Receptor. In Heterologous Expression of Membrane Proteins; Springer: New York, NY, USA, 2022; Volume 2507, pp. 313–325.
- Darrouzet, E.; Lindenthal, S.; Marcellin, D.; Pellequer, J.-L.; Pourcher, T. The sodium/iodide symporter: State of the art of its molecular characterization. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838 Pt B, 244–253.
- Ravera, S.; Reyna-Neyra, A.; Ferrandino, G.; Amzel, L.M.; Carrasco, N. The Sodium/Iodide Symporter (NIS): Molecular Physiology and Preclinical and Clinical Applications. Annu. Rev. Physiol. 2017, 79, 261–289.
- Riesco-Eizaguirre, G.; Santisteban, P.; De la Vieja, A. The complex regulation of NIS expression and activity in thyroid and extrathyroidal tissues. Endocrine-Related Cancer 2021, 28, T141–T165.
- Thompson, R.J.; Fletcher, A.; Brookes, K.; Nieto, H.; Alshahrani, M.M.; Mueller, J.W.; Fine, N.H.; Hodson, D.J.; Boelaert, K.; Read, M.; et al. Dimerization of the Sodium/Iodide Symporter. Thyroid 2019, 29, 1485–1498.
- Rozenfeld, J.; Efrati, E.; Adler, L.; Tal, O.; Carrithers, S.L.; Alper, S.L.; Zelikovic, I. Transcriptional Regulation of the Pendrin Gene. Cell. Physiol. Biochem. 2011, 28, 385–396.
- Dossena, S.; Nofziger, C.; Tamma, G.; Bernardinelli, E.; Vanoni, S.; Nowak, C.; Grabmayer, E.; Kössler, S.; Stephan, S.; Patsch, W.; et al. Molecular and Functional Characterization of Human Pendrin and its Allelic Variants. Cell. Physiol. Biochem. 2011, 28, 451–466.
- Dossena, S.; Bizhanova, A.; Nofziger, C.; Bernardinelli, E.; Ramsauer, J.; Kopp, P.; Paulmichl, M. Identification of Allelic Variants of Pendrin (SLC26A4) with Loss and Gain of Function. Cell. Physiol. Biochem. 2011, 28, 467–476.
- Bizhanova, A.; Kopp, P. Controversies Concerning the Role of Pendrin as an Apical Iodide Transporter in Thyroid Follicular Cells. Cell. Physiol. Biochem. 2011, 28, 485–490.
- Twyffels, L.; Massart, C.; Golstein, P.E.; Raspe, E.; Van Sande, J.; Dumont, J.E.; Beauwens, R.; Kruys, V. Pendrin: The Thyrocyte Apical Membrane Iodide Transporter? Cell. Physiol. Biochem. 2011, 28, 491–496.
- Zheng, G.; Marino, M.; Zhao, J.; McCluskey, R.T. Megalin (gp330): A Putative Endocytic Receptor for Thyroglobulin (Tg). Endocrinology 1998, 139, 1462–1465.
- Marinò, M.; Pinchera, A.; McCluskey, R.T.; Chiovato, L. Megalin in Thyroid Physiology and Pathology. Thyroid 2001, 11, 47–56.
- Marinò, M.; Zheng, G.; Chiovato, L.; Pinchera, A.; Brown, D.; Andrews, D.; McCluskey, R.T. Role of Megalin (gp330) in Transcytosis of Thyroglobulin by Thyroid Cells. A novel function in the control of thyroid hormone release. J. Biol. Chem. 2000, 275, 7125–7137.
- Soh, S.-B.; Aw, T.-C. Laboratory Testing in Thyroid Conditions—Pitfalls and Clinical Utility. Ann. Lab. Med. 2019, 39, 3–14.
- Doggui, R. Immunoanalytical profile of thyroglobulin antibodies. Ann. Biol. Clin. 2018, 76, 695–704.
- Dwivedi, S.N.; Kalaria, T.; Buch, H. Thyroid autoantibodies. J. Clin. Pathol. 2023, 76, 19–28.
- McLeod, D.S.A.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265.
- McLachlan, S.M.; Rapoport, B. Thyroid Autoantibodies Display both “Original Antigenic Sin” and Epitope Spreading. Front. Immunol. 2017, 8, 1845.
- Baker, S.; Miguel, R.N.; Thomas, D.; Powell, M.; Furmaniak, J.; Smith, B.R. Cryo-electron microscopy structures of human thyroid peroxidase (TPO) in complex with TPO antibodies. J. Mol. Endocrinol. 2023, 70, e220149.
- Espenbetova, M.; Kuzmina, N.; Zubkov, A.; Akhmetova, V.; Zamanbekova, Z.; Krykpaeva, A.; Zhumanbayeva, Z.; Amrenova, K.; Smailova, Z.; Glushkova, N. Epitopes specificity of antibodies to thyroid peroxidase in patients with Graves’ disease, Hashimoto’s thyroiditis and overlap-syndrome. J. Clin. Transl. Endocrinol. 2022, 27, 100293.
- Ehlers, M.; Allelein, S.; Schott, M. TSH-receptor autoantibodies: Pathophysiology, assay methods, and clinical applications. Minerva Endocrinol. 2018, 43, 323–332.
- Nicolì, F.; Lanzolla, G.; Mantuano, M.; Ionni, I.; Mazzi, B.; Leo, M.; Sframeli, A.; Posarelli, C.; Maglionico, M.N.; Figus, M.; et al. Correlation between serum anti-TSH receptor autoantibodies (TRAbs) and the clinical feature of Graves’ orbitopathy. J. Endocrinol. Investig. 2021, 44, 581–585.
- Kahaly, G.J.; Diana, T.; Olivo, P.D. TSH Receptor Antibodies: Relevance & Utility. Endocr. Pract. 2020, 26, 97–106.
- Shrestha, A.; Adhikari, N.; Devkota, S.; Chowdhury, T.; Shiferaw-Deribe, Z.; Gousy, N.; Adhikari, S. Fluctuating Hyperthyroidism and Hypothyroidism in Graves’ Disease: The Swinging Between Two Clinical Entities. Cureus 2022, 14, e27715.
- Santos, T.D.S.; Oliveira, J.C.; Freitas, C.; de Carvalho, A.C. Thyroid-Stimulatory Antibody as a Predictive Factor for Graves’ Disease Relapse. Cureus 2022, 14, e22190.
- Arshad, I.; Zahra, T.; Vargas-Jerez, J. New-Onset Graves’ Disease in the Background of Hashimoto’s Thyroiditis: Spectrums of the Same Disease With Changing Autoantibodies. Cureus 2022, 14, e28296.
- Kotwal, A.; Stan, M. Thyrotropin Receptor Antibodies—An Overview. Ophthalmic Plast. Reconstr. Surg. 2018, 34 (Suppl. S1), S20–S27.
- Michalek, K.; Morshed, S.A.; Latif, R.; Davies, T.F. TSH receptor autoantibodies. Autoimmun. Rev. 2009, 9, 113–116.
- Brix, T.H.; Hegedüs, L.; Weetman, A.P.; Kemp, H.E. Pendrin and NIS antibodies are absent in healthy individuals and are rare in autoimmune thyroid disease: Evidence from a Danish twin study. Clin. Endocrinol. 2014, 81, 440–444.
- Eleftheriadou, A.-M.; Mehl, S.; Renko, K.; Kasim, R.H.; Schaefer, J.-A.; Minich, W.B.; Schomburg, L. Re-visiting autoimmunity to sodium-iodide symporter and pendrin in thyroid disease. Eur. J. Endocrinol. 2020, 183, 571–580.
- Lisi, S.; Pinchera, A.; McCluskey, R.T.; Willnow, T.E.; Refetoff, S.; Marcocci, C.; Vitti, P.; Menconi, F.; Grasso, L.; Luchetti, F.; et al. Preferential megalin-mediated transcytosis of low-hormonogenic thyroglobulin: A control mechanism for thyroid hormone release. Proc. Natl. Acad. Sci. USA 2003, 100, 14858–14863.
- Wang, Y.M.; Lee, V.W.; Wu, H.; Harris, D.C.; Alexander, S.I. Heymann Nephritis in Lewis Rats. Curr. Protoc. Immunol. 2015, 109, 15.29.1–15.29.6.
- Akiyama, S.; Imai, E.; Maruyama, S. Immunology of membranous nephropathy. F1000Research 2019, 8, 734.
- Marinoò, M.; Chiovato, L.; Friedlander, J.A.; Latrofa, F.; Pinchera, A.; McCluskey, R.T. Serum Antibodies against Megalin (GP330) in Patients with Autoimmune Thyroiditis1. J. Clin. Endocrinol. Metab. 1999, 84, 2468–2474.
- Esfandiari, N.H.; Papaleontiou, M. Biochemical Testing in Thyroid Disorders. Endocrinol. Metab. Clin. N. Am. 2017, 46, 631–648.
- Fröhlich, E.; Wahl, R. Thyroid Autoimmunity: Role of Anti-thyroid Antibodies in Thyroid and Extra-Thyroidal Diseases. Front. Immunol. 2017, 8, 521.
- Vargas-Uricoechea, H.; Agredo-Delgado, V.; Vargas-Sierra, H.D.; Pinzón-Fernández, M.V. Prevalence of Functional Alterations and the Effects of Thyroid Autoimmunity on the Levels of TSH in an Urban Population of Colombia: A Population-Based Study. Endocr. Metab. Immune Disord. Drug Targets 2022, 23, 857–866.
- Czarnocka, B. Thyroperoxidase, thyroglobulin, Na+/I- symporter, pendrin in thyroid autoimmunity. Front. Biosci. 2011, 16, 783–802.
- Hadj-Kacem, H.; Rebuffat, S.; Mnif-Féki, M.; Belguith-Maalej, S.; Ayadi, H.; Péraldi-Roux, S. Autoimmune thyroid diseases: Genetic susceptibility of thyroid-specific genes and thyroid autoantigens contributions. Int. J. Immunogenetics 2009, 36, 85–96.
- Baloch, Z.; Carayon, P.; Conte-Devolx, B.; Demers, L.M.; Feldt-Rasmussen, U.; Henry, J.-F.; LiVosli, V.A.; Niccoli-Sire, P.; John, R.; Ruf, J.; et al. Laboratory Support for the Diagnosis and Monitoring of Thyroid Disease. Thyroid 2003, 13, 3–126.
- Demers, L.M.; Spencer, C.A. Laboratory medicine practice guidelines: Laboratory support for the diagnosis and monitoring of thyroid disease. Clin. Endocrinol. 2003, 58, 138–140.
- Furmaniak, J.; Sanders, J.; Sanders, P.; Miller-Gallacher, J.; Ryder, M.M.; Smith, B.R. Practical applications of studies on the TSH receptor and TSH receptor autoantibodies. Endocrine 2020, 68, 261–264.
- Kahaly, G.J.; Bartalena, L.; Hegedüs, L.; Leenhardt, L.; Poppe, K.; Pearce, S.H. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur. Thyroid. J. 2018, 7, 167–186.
- Parameswaran, R.; de Jong, M.C.; Kit, J.L.W.; Sek, K.; Nam, T.Q.; Thang, T.V.; Khue, N.T.; Aye, T.T.; Tun, P.M.; Cole, T.; et al. 2021 Asia-Pacific Graves’ Disease Consortium Survey of Clinical Practice Patterns in the Management of Graves’ Disease. Endocrine 2023, 79, 135–142.
- Hoang, T.D.; Stocker, D.J.; Chou, E.L.; Burch, H.B. 2022 Update on Clinical Management of Graves Disease and Thyroid Eye Disease. Endocrinol. Metab. Clin. N. Am. 2022, 51, 287–304.
- Benvenga, S.; Guarneri, F. Homology of pendrin, sodium-iodide symporter and apical iodide transporter. Front. Biosci. Landmark Ed. 2018, 23, 1864–1873.
- Benvenga, S.; Bartolone, L.; Squadrito, S.; Trimarchi, F. Thyroid Hormone Autoantibodies Elicited by Diagnostic Fine Needle Biopsy 1. J. Clin. Endocrinol. Metab. 1997, 82, 4217–4223.
- Benvenga, S.; Burek, C.L.; Talor, M.; Rose, N.R.; Trimarchi, F. Heterogeneity of the thyroglobulin epitopes associated with circulating thyroid hormone autoantibodies in Hashimoto’s thyroiditis and non-autoimmune thyroid diseases. J. Endocrinol. Investig. 2002, 25, 977–982.
- Sakata, S.; Nakamura, S.; Miura, K. Autoantibodies against thyroid hormones or iodothyronines. Ann. Intern. Med. 1985, 103, 579–589.
- Ni, J.; Long, Y.; Zhang, L.; Yang, Q.; Kou, C.; Li, S.; Li, J.; Zhang, H. High prevalence of thyroid hormone autoantibody and low rate of thyroid hormone detection interference. J. Clin. Lab. Anal. 2022, 36, e24124.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
668
Revisions:
2 times
(View History)
Update Date:
25 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No