Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yilun Liu | -- | 1554 | 2023-08-22 05:42:57 | | | |
| 2 | Wendy Huang | Meta information modification | 1554 | 2023-08-24 09:43:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mo, C.; Shiozaki, Y.; Omabe, K.; Liu, Y. RECQ5 Functions in DNA Repair and Transcription. Encyclopedia. Available online: https://encyclopedia.pub/entry/48298 (accessed on 27 July 2026).
Mo C, Shiozaki Y, Omabe K, Liu Y. RECQ5 Functions in DNA Repair and Transcription. Encyclopedia. Available at: https://encyclopedia.pub/entry/48298. Accessed July 27, 2026.
Mo, Chiefe, Yukari Shiozaki, Kenneth Omabe, Yilun Liu. "RECQ5 Functions in DNA Repair and Transcription" Encyclopedia, https://encyclopedia.pub/entry/48298 (accessed July 27, 2026).
Mo, C., Shiozaki, Y., Omabe, K., & Liu, Y. (2023, August 22). RECQ5 Functions in DNA Repair and Transcription. In Encyclopedia. https://encyclopedia.pub/entry/48298
Mo, Chiefe, et al. "RECQ5 Functions in DNA Repair and Transcription." Encyclopedia. Web. 22 August, 2023.
Copy Citation
RECQ5, a member of the conserved RECQ helicase family, is the sole human RECQ homolog that has not been linked to a hereditary developmental syndrome. Nonetheless, dysregulation of RECQ5 has emerged as a significant clinical concern, being linked to cancer predisposition, cardiovascular disease, and inflammation. In cells, RECQ5 assumes a crucial role in the regulation of DNA repair pathways, particularly in the repair of DNA double-strand breaks and inter-strand DNA crosslinks. Moreover, RECQ5 exhibits a capacity to modulate gene expression by interacting with transcription machineries and their co-regulatory proteins, thus safeguarding against transcription-induced DNA damage.
RECQ5
transcription
replication
DNA repair
helicase
1. Introduction
The RECQ family of DNA helicases is present in both prokaryotes and eukaryotes [1]. In humans, five RECQ homologs have been identified: RECQL/RECQ1, BLM/RECQ2, WRN/RECQ3, RECQ4, and RECQ5. Although there are multiple RECQ helicases in human cells, it is important to note that their sequence homologies are limited to the helicase domain [1]. This limited sequence similarity explains why mutations in different RECQ genes are associated with distinct diseases and why the clinical manifestation of those mutations in a particular RECQ helicase cannot be compensated for by the presence of other RECQ homologs.
The clinical significance of the RECQ family of DNA helicases was recognized long before the identification of the five RECQ genes in humans. In 1868, Dr. August Rothmund described the first RECQ-associated disease called Rothmund–Thomson syndrome (RTS), characterized by developmental abnormalities, premature aging, and cancer [1][2]. However, it took another 120 years to establish the connection between RTS and mutations in the RECQ4 gene [3]. In 1904, Dr. Otto Werner reported a progeria syndrome in his doctoral dissertation, which would later be associated with WRN/RECQ3 and named Werner’s syndrome [4]. In 1954, another clinical case of congenital telangiectatic erythema with short stature was presented by Dr. David Bloom in New York, leading to the identification of Bloom’s syndrome [5][6]. The underlying cause of Bloom’s syndrome was revealed in 1965 when Dr. Bloom, in collaboration with Dr. James German, observed highly unstable chromosomes with increased DNA breakages, sister chromatin exchanges, and a quadriradial configuration in patients with Bloom’s syndrome [7]. The BLM gene, responsible for Bloom’s syndrome, was cloned in 1995 and found to share sequence similarities with the RECQ family of DNA helicases, including RECQL/RECQ1 [8][9][10]. The third RECQ gene, initially known as RECQ3, was isolated during the search for gene mutations underlying Werner’s syndrome and later renamed WRN [11].
In 1998, the RECQ4 and RECQ5 genes, also known as RECQL4 and RECQL5, respectively, were successfully isolated through a sequence homology search targeting the known RECQ family helicases [12]. Building upon the associations of BLM/RECQ2 and WRN/RECQ3 with Bloom’s syndrome and Werner’s syndrome, investigations were initiated to uncover hereditary diseases linked to RECQ1, RECQ4, and RECQ5. In 1999, Dr. Yasuhiro Furuichi and his team at the AGENE Research Institute in Japan made a significant breakthrough by establishing the connection between mutations in the RECQ4 gene and a specific subset of RTS [3]. A few years later, RECQ4 mutations were also found to be associated with two additional developmental disorders, namely RAPADILINO syndrome and Baller–Gerold syndrome [13][14]. Another clinical milestone was recently reached, when mutations in the RECQ1 gene were identified as the cause of the RECQL ONE (RECON) syndrome, characterized by a short stature, premature facial aging, xeroderma, and disproportional finger lengths [15]. Together, these findings have significantly contributed to the understanding of the clinical spectrum and genetic bases of RECQ helicase-related disorders, shedding light on the intricate roles played by these genes in human health and development.
2. RECQ5 Functions in DNA Repair
The RECQ5 gene has been identified in various multicellular organisms, including Caenorhabditis elegans (C. elegans) [16], Drosophila [17][18], mice [19], and chicken [20]. This allows for the study of RECQ5 deficiency in different model organisms, providing valuable insights even in the absence of direct links to human diseases. In C. elegans, although RECQ5 deficiency does not impact development, it does result in a shortened lifespan [21]. Because C. elegans RECQ5 is ubiquitously expressed and particularly enriched in the intestine compared to other organs, it is possible that in the absence of RECQ5, the accumulation of DNA damage in the intestine resulting from oxidative stress caused by environmental insults and food metabolism contributes to a shortened lifespan [21]. Similarly, in a mouse model with the deletion of the Recq5 gene, no developmental abnormalities were observed, but these mice exhibited a strikingly high susceptibility to cancer at an older age compared to their wild-type counterparts [22]. At the cellular level, Recq5 knockout (KO) mouse embryonic stem cells display an increase in sister chromatid exchanges (SCEs) due to an elevated frequency of homologous recombination (HR) in repairing DNA double-stranded breaks (DSBs) [22], resembling Dr. German’s initial observation in cells derived from Bloom’s syndrome patients [7]. Interestingly, in chicken DT40 cells, RECQ5 deletion alone does not lead to significant increases in SCEs. However, when combined with a BLM−/− mutation, RECQ5-BLM double-mutant cells exhibit significantly higher SCE levels compared to BLM single-mutant or RECQ5 single-mutant cells. This suggests that RECQ5 functions as a backup for BLM in suppressing SCEs in chicken cells [20]. Nonetheless, several studies support the notion of a non-redundant role of RECQ5 in DNA damage repair, at least in mammalian cells [22][23][24].
RAD51 is essential for the strand invasion and pairing step of HR, and RECQ5 plays a crucial role in limiting SCEs by disrupting RAD51 presynaptic filaments during the initiation step of HR [22]. This activity is supported by the observation that RECQ5 localizes to DSBs [25][26], and its expression is inversely correlated with the number of RAD51 foci per cell [22]. The anti-RAD51 recombination function of RECQ5 is mediated through a direct interaction between RAD51 and the unique C-terminus of RECQ5 [27]. Sequence analysis has revealed that this RAD51-interacting domain of RECQ5, spanning residues 662 to 706, contains a BRC repeat motif like those found in breast cancer gene 2 (BRCA2), which interacts with RAD51 to promote HR in DSB repair [28]. Single-molecule studies have indicated that RECQ5 translocates along the RAD51 presynaptic filament in an ATP-dependent manner. This movement is believed to facilitate the disassembly of the filament and to limit the subsequent steps such as strand invasion and pairing to form a D-loop structure [29]. Additionally, by removing RAD51 from stalled replication forks, RECQ5 promotes access to the MUS81-EME1 flap endonuclease, which cleaves the stalled replication fork to resolve it [30]. Importantly, the dissociation of RAD51 filaments by RECQ5 also directs the repair of DSBs toward the synthesis-dependent strand annealing (SDSA) pathway that results in a noncrossover or alternative homology-dependent repair (HDR) utilizing a ssDNA donor independent of RAD51 [31][32].
3. RECQ5 Function in Transcription
In 1998, when the RECQ5 gene was cloned, researchers initially expected it to be mainly involved in DNA repair based on the sequence homology to other RECQ homologs. It was, therefore, a surprise when RECQ5 was also linked to transcription regulation. Specifically, RECQ5 has been identified as an RNA polymerase II (RNAPII)-associated protein [33][34][35]. Through domain mapping, two regions located at the C-terminal portion of the RECQ5 protein were identified as being responsible for the direct interaction with RPB1, the largest subunit of RNAPII [33][36]. Sequence analysis revealed that these two RNAPII interaction domains share sequence homology with the KIX domain and SET2-Rpb1 interacting (SRI) domain found in transcription factors CBP and SET2, respectively (Figure 1) [36]. The KIX domain primarily interacts with RNAPIIa, the inactive and non-phosphorylated form of RNAPII, while the SRI domain is critical for the interaction with active, hyperphosphorylated RNAPIIo [34][36][37][38]. This distinct preference in interactions with different forms of RNAPII allows RECQ5 to function as both a negative and positive regulator of RNAPII-dependent transcription. The binding of RECQ5 via the KIX domain inhibits RNAPII at both the initiation and elongation steps in in vitro transcriptional assays, and this inhibition does not require RECQ5 ATPase activity [39]. Structural analysis suggests that the KIX domain binds to RNAPII in a manner similar to that of the TFIIS transcription factor, which is important for transcription elongation, thereby sterically blocking transcription elongation and preventing TFIIS interaction with RNAPII [40]. Given that the KIX domain overlaps with residues 561–651, which are responsible for single-stranded annealing activity [41][42], it would be worth investigating if RECQ5’s annealing activity contributes to transcriptional inhibition by preventing ssDNA formation at the promoter region, thereby reducing promoter accessibility to RNAPII.
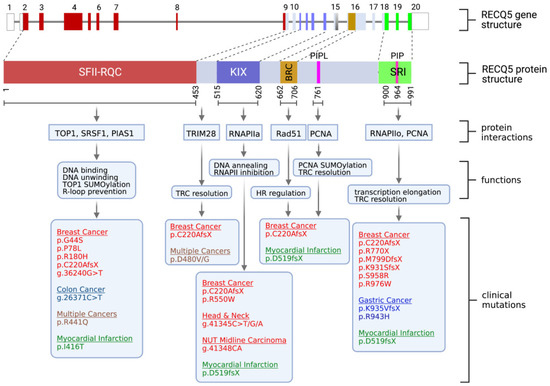
Figure 1. Summary of human RECQ5 gene structure, RECQ5 protein domains, biochemical properties, cellular functions, and RECQ5 clinical variants. (Top) Gene structure of the human RECQ5 gene, including exons and introns, and the RECQ5 protein domains, including the Superfamily helicase II (SFII), RECQ-C-terminus (RQC), KIX, BRC, PIP-L, PIP and SRI domains. (Middle) Summary of the biochemical properties and functions of the RECQ5 domains. (Bottom) RECQ5 variants that have been linked to cancer or cardiovascular disease.
In addition to its negative regulation of RNAPII-dependent transcription via the KIX domain, several studies support the role of RECQ5 during transcription elongation through the SRI domain [38]. RNAPII-dependent transcription is initiated by the TFIIH-mediated Ser5 phosphorylation of the RNAPII C-terminal repeat domain (CTD), followed by the Ser2 phosphorylation of the CTD as RNAPII transitions into the elongation mode [43]. The SRI domain of RECQ5 binds to hyperphosphorylated RNAPIIo proteins at both Ser2 and Ser5, and this binding correlates with the RECQ5 association with gene regions that are being actively transcribed [38]. One key function of RECQ5 in transcription elongation is to control the speed of RNAPIIo to prevent transcription stalling and backtracking [44].
References
- Liu, Y. Rothmund-Thomson syndrome helicase, RECQ4: On the crossroad between DNA replication and repair. DNA Repair 2010, 9, 325–330.
- Rothmund, A. Ueber Cataracten in Verbindung mit einer eigenthümlichen Hautdegeneration. Arch. Ophthalmol. 1868, 14, 159–182.
- Kitao, S.; Shimamoto, A.; Goto, M.; Miller, R.W.; Smithson, W.A.; Lindor, N.M.; Furuichi, Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999, 22, 82–84.
- On cataract in conjunction with scleroderma. Otto Werner, doctoral dissertation, 1904, Royal Ophthalmology Clinic, Royal Christian Albrecht University of Kiel. Adv. Exp. Med. Biol. 1985, 190, 1–14.
- Bloom, D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am. J. Dis. Child. 1954, 88, 754–758.
- Passarge, E. James L. German, a pioneer in early human genetic research turned 90. Am. J. Med. Genet. A 2016, 170, 1564–1565.
- German, J.; Archibald, R.; Bloom, D. Chromosomal Breakage in a Rare and Probably Genetically Determined Syndrome of Man. Science 1965, 148, 506–507.
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666.
- Puranam, K.L.; Blackshear, P.J. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J. Biol. Chem. 1994, 269, 29838–29845.
- Seki, M.; Miyazawa, H.; Tada, S.; Yanagisawa, J.; Yamaoka, T.; Hoshino, S.; Ozawa, K.; Eki, T.; Nogami, M.; Okumura, K.; et al. Molecular cloning of cDNA encoding human DNA helicase Q1 which has homology to Escherichia coli Rec Q helicase and localization of the gene at chromosome 12p12. Nucleic Acids Res. 1994, 22, 4566–4573.
- Yu, C.E.; Oshima, J.; Fu, Y.H.; Wijsman, E.M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Ouais, S.; et al. Positional cloning of the Werner’s syndrome gene. Science 1996, 272, 258–262.
- Kitao, S.; Ohsugi, I.; Ichikawa, K.; Goto, M.; Furuichi, Y.; Shimamoto, A. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics 1998, 54, 443–452.
- Siitonen, H.A.; Kopra, O.; Kääriäinen, H.; Haravuori, H.; Winter, R.M.; Säämänen, A.M.; Peltonen, L.; Kestilä, M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 2003, 12, 2837–2844.
- Van Maldergem, L.; Siitonen, H.A.; Jalkh, N.; Chouery, E.; De Roy, M.; Delague, V.; Muenke, M.; Jabs, E.W.; Cai, J.; Wang, L.L.; et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 2006, 43, 148–152.
- Datta, A.; Sommers, J.A.; Jhujh, S.S.; Harel, T.; Stewart, G.S.; Brosh, R.M., Jr. Discovery of a new hereditary RECQ helicase disorder RECON syndrome positions the replication stress response and genome homeostasis as centrally important processes in aging and age-related disease. Ageing Res. Rev. 2023, 86, 101887.
- Wilson, R.; Ainscough, R.; Anderson, K.; Baynes, C.; Berks, M.; Bonfield, J.; Burton, J.; Connell, M.; Copsey, T.; Cooper, J.; et al. 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature 1994, 368, 32–38.
- Sekelsky, J.J.; Brodsky, M.H.; Rubin, G.M.; Hawley, R.S. Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res. 1999, 27, 3762–3769.
- Jeong, S.M.; Kawasaki, K.; Juni, N.; Shibata, T. Identification of Drosophila melanogaster RECQE as a member of a new family of RecQ homologues that is preferentially expressed in early embryos. Mol. Gen. Genet. 2000, 263, 183–193.
- Ohhata, T.; Araki, R.; Fukumura, R.; Kuroiwa, A.; Matsuda, Y.; Abe, M. Cloning, genomic structure and chromosomal localization of the gene encoding mouse DNA helicase RECQL5beta. Gene 2001, 280, 59–66.
- Wang, W.; Seki, M.; Narita, Y.; Nakagawa, T.; Yoshimura, A.; Otsuki, M.; Kawabe, Y.-I.; Tada, S.; Yagi, H.; Ishii, Y.; et al. Functional Relation among RecQ Family Helicases RecQL1, RecQL5, and BLM in Cell Growth and Sister Chromatid Exchange Formation. Mol. Cell. Biol. 2003, 23, 3527–3535.
- Jeong, Y.S.; Kang, Y.L.; Lim, K.H.; Lee, M.H.; Lee, J.; Koo, H.-S. Deficiency of Caenorhabditis elegans RecQ5 homologue reduces life span and increases sensitivity to ionizing radiation. DNA Repair 2003, 2, 1309–1319.
- Hu, Y.; Raynard, S.; Sehorn, M.G.; Lu, X.; Bussen, W.; Zheng, L.; Stark, J.M.; Barnes, E.L.; Chi, P.; Janscak, P.; et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007, 21, 3073–3084.
- Hu, Y.; Lu, X.; Barnes, E.; Yan, M.; Lou, H.; Luo, G. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol. Cell. Biol. 2005, 25, 3431–3442.
- Kim, T.M.; Son, M.Y.; Dodds, S.; Hu, L.; Luo, G.; Hasty, P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015, 43, 893–903.
- Popuri, V.; Huang, J.; Ramamoorthy, M.; Tadokoro, T.; Croteau, D.L.; Bohr, V.A. RECQL5 plays co-operative and complementary roles with WRN syndrome helicase. Nucleic Acids Res. 2017, 45, 1566.
- Ramamoorthy, M.; May, A.; Tadokoro, T.; Popuri, V.; Seidman, M.M.; Croteau, D.L.; Bohr, V.A. The RecQ helicase RECQL5 participates in psoralen-induced interstrand cross-link repair. Carcinogenesis 2013, 34, 2218–2230.
- Schwendener, S.; Raynard, S.; Paliwal, S.; Cheng, A.; Kanagaraj, R.; Shevelev, I.; Stark, J.M.; Sung, P.; Janscak, P. Physical Interaction of RECQ5 Helicase with RAD51 Facilitates Its Anti-recombinase Activity. J. Biol. Chem. 2010, 285, 15739–15745.
- Islam, M.N.; Paquet, N.; Fox, D., 3rd; Dray, E.; Zheng, X.F.; Klein, H.; Sung, P.; Wang, W. A variant of the breast cancer type 2 susceptibility protein (BRC) repeat is essential for the RECQL5 helicase to interact with RAD51 recombinase for genome stabilization. J. Biol. Chem. 2012, 287, 23808–23818.
- Xue, C.; Molnarova, L.; Steinfeld, J.B.; Zhao, W.; Ma, C.; Spirek, M.; Kaniecki, K.; Kwon, Y.; Beláň, O.; Krejci, K.; et al. Single-molecule visualization of human RECQ5 interactions with single-stranded DNA recombination intermediates. Nucleic Acids Res. 2021, 49, 285–305.
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Hühn, D.; et al. RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol. Cell 2017, 66, 658–671.e8.
- Paliwal, S.; Kanagaraj, R.; Sturzenegger, A.; Burdova, K.; Janscak, P. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res. 2014, 42, 2380–2390.
- Olson, H.C.; Davis, L.; Kiianitsa, K.; Khoo, K.J.; Liu, Y.; Knijnenburg, T.A.; Maizels, N. Increased levels of RECQ5 shift DNA repair from canonical to alternative pathways. Nucleic Acids Res. 2018, 46, 9496–9509.
- Aygün, O.; Svejstrup, J.; Liu, Y. A RECQ5–RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. USA 2008, 105, 8580–8584.
- Izumikawa, K.; Yanagida, M.; Hayano, T.; Tachikawa, H.; Komatsu, W.; Shimamoto, A.; Futami, K.; Furuichi, Y.; Shinkawa, T.; Yamauchi, Y.; et al. Association of human DNA helicase RecQ5beta with RNA polymerase II and its possible role in transcription. Biochem. J. 2008, 413, 505–516.
- Zhou, G.; Liu, Y.; Wu, S.Y.; Tie, F.; Lou, H.; Chiang, C.M.; Luo, G. Purification of a novel RECQL5-SWI/SNF-RNAPII super complex. Int. J. Biochem. Mol. Biol. 2010, 1, 101–111.
- Islam, M.N.; Fox, D.; Guo, R.; Enomoto, T.; Wang, W. RecQL5 Promotes Genome Stabilization through Two Parallel Mechanisms—Interacting with RNA Polymerase II and Acting as a Helicase. Mol. Cell. Biol. 2010, 30, 2460–2472.
- Li, M.; Xu, X.; Liu, Y. The SET2-RPB1 interaction domain of human RECQ5 is important for transcription-associated genome stability. Mol. Cell. Biol. 2011, 31, 2090–2099.
- Kanagaraj, R.; Huehn, D.; MacKellar, A.; Menigatti, M.; Zheng, L.; Urban, V.; Shevelev, I.; Greenleaf, A.L.; Janscak, P. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010, 38, 8131–8140.
- Aygün, O.; Xu, X.; Liu, Y.; Takahashi, H.; Kong, S.E.; Conaway, R.C.; Conaway, J.W.; Svejstrup, J.Q. Direct inhibition of RNA polymerase II transcription by RECQL5. J. Biol. Chem. 2009, 284, 23197–23203.
- Kassube, S.A.; Jinek, M.; Fang, J.; Tsutakawa, S.; Nogales, E. Structural mimicry in transcription regulation of human RNA polymerase II by the DNA helicase RECQL5. Nat. Struct. Mol. Biol. 2013, 20, 892–899.
- Garcia, P.L.; Liu, Y.; Jiricny, J.; West, S.C.; Janscak, P. Human RECQ5beta, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 2004, 23, 2882–2891.
- Kanagaraj, R.; Saydam, N.; Garcia, P.L.; Zheng, L.; Janscak, P. Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 2006, 34, 5217–5231.
- Phatnani, H.P.; Greenleaf, A.L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006, 20, 2922–2936.
- Saponaro, M.; Kantidakis, T.; Mitter, R.; Kelly, G.P.; Heron, M.; Williams, H.; Söding, J.; Stewart, A.; Svejstrup, J.Q. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014, 157, 1037–1049.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
661
Revisions:
2 times
(View History)
Update Date:
24 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No