Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jenchin Wang | -- | 2953 | 2023-08-21 02:27:40 | | | |
| 2 | Camila Xu | Meta information modification | 2953 | 2023-08-21 04:57:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yadav, R.; Hakobyan, N.; Wang, J. Mechanism of Action of Lymphocyte Activation Gene 3. Encyclopedia. Available online: https://encyclopedia.pub/entry/48251 (accessed on 28 July 2026).
Yadav R, Hakobyan N, Wang J. Mechanism of Action of Lymphocyte Activation Gene 3. Encyclopedia. Available at: https://encyclopedia.pub/entry/48251. Accessed July 28, 2026.
Yadav, Ruchi, Narek Hakobyan, Jen-Chin Wang. "Mechanism of Action of Lymphocyte Activation Gene 3" Encyclopedia, https://encyclopedia.pub/entry/48251 (accessed July 28, 2026).
Yadav, R., Hakobyan, N., & Wang, J. (2023, August 21). Mechanism of Action of Lymphocyte Activation Gene 3. In Encyclopedia. https://encyclopedia.pub/entry/48251
Yadav, Ruchi, et al. "Mechanism of Action of Lymphocyte Activation Gene 3." Encyclopedia. Web. 21 August, 2023.
Copy Citation
Lymphocyte activation gene 3 (LAG-3) (CD223) is a CD4-related activation-induced cell surface inhibitory receptor that binds to major histocompatibility complex (MHC) class II molecules with high affinity and negatively regulates T cell effector functions.
Myeloproliferative neoplasm
Immune checkpoint inhibitor
Philadelphia chromosome
1. Introduction
The myeloproliferative neoplasms (MPN), are characterized by the clonal proliferation of one or more hematopoietic cell lineages [1]. As per International Consensus Classification [2] (ICC) and World Health Organization (WHO), 5th edition [3] classic MPN comprises mainly essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF) [4]. Recently, there has been a notable shift in the treatment approach for MPN, with an increasing emphasis on agents that specifically target the immune system, cytokine milieu, immunomodulatory agents, and targeted immune therapy. The pathogenesis of MPN is not very clear but studies have shown that TNF-α selectively promotes the growth of JAK2V617F-positive MPN cells over controls contributing to clonal expansion of mutant copies during MPN progression [5]. Various driver mutations as studied by genetic sequencing, clonal analysis, and protein expression showed that tyrosine kinase Janus Kinase 2 -JAK2V617F mutation occurs in 95% of patients with PV and 50% to 60% of patients with ET and MF [6][7]. These mutations lead to hyperactive Janus kinase, signal transducer, and activator of transcription proteins (JAK-STAT) signaling pathways downstream of the erythropoietin receptor and thrombopoietin receptor (MPL) [8]. Concurrently profound immune dysregulation and defective immune surveillance also play a significant role in the pathogenesis of MPN [9]. The dysregulated genes associated with the immune system and inflammation, which have been implicated in myeloproliferative neoplasms (MPN), include the interferon-inducible gene [10], regulatory T cells (such as CD4+ CD25+ FOXP3+ Tregs) [11], natural killer cells, human leukocyte antigen (HLA) class I and II molecules, β2-microglobulin, molecules involved in the processing of HLA I antigens (such as LMP2, LMP7, TAP1/2, and tapasin) [10][12], as well as antioxidative stress genes (ATM, TP53, CYBA, NRF2, PTGS1, and SIRT2) [13][14]. Furthermore, the upregulation of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), contributes to the evasion of tumor cells from immune surveillance, thus playing a crucial role in the etiopathogenesis of MPN [15]. The key events involved in the development of the neoplastic process are oncogenic transformation and immune escape allowing for uncontrolled proliferation and avoidance of apoptosis. Immune checkpoint inhibitory therapy (ICIT) is based on blocking the inhibitory pathways in T cells to promote anti-tumor immune responses and has revolutionized cancer treatment paradigms. Oncogenic JAK2 activation results in high expression of programmed death-ligand 1 (PD-L1) on the surface of monocytes, megakaryocytes, MDSC, and platelets which is mediated via the JAK2-STAT3 and JAK2-STAT5 axes [16]. The blocking of the PD-1 pathway, which was found to be overexpressed in myeloid malignancies, has gained immense interest as a therapeutic target paving the path for novel strategies. One such trial was ClinicalTrials.gov Identifier: NCT02421354 where the efficacy and safety of single-agent nivolumab (PD-1 inhibitor) in eight adult patients with myelofibrosis was tested [17]. However, this study was terminated early due to failure to meet the predetermined efficacy endpoint. At the American Society of Hematology (ASH) annual meeting in 2020, a multi-center, open-label, Phase II, single-arm study of pembrolizumab was presented with its use in patients with primary, post-essential thrombocythemia or post-polycythemia vera MF (NCT03065400) [18]. Nine cases were presented, but none had a clinical response.
The use of ICI in hematological malignancies brings a daunting challenge with a low response rate thus letting the oncologist/molecular physicians change the focus to dig deeply into the tumor microenvironment for alternative therapeutic targets. To permit more patients to benefit from immunotherapy, the focus has changed to targeting alternative novel immune checkpoints in the tumor microenvironment such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin, mucin domain 3 (TIM-3), T cell immunoglobulin, ITIM domain (TIGIT), V-domain immunoglobulin-containing suppressor of T cell activation (VISTA) and human endogenous retrovirus-H long terminal repeat-associating protein 2 (HHLA2) forming the basis of next-generation ICIT [19] as shown in Figure 1.
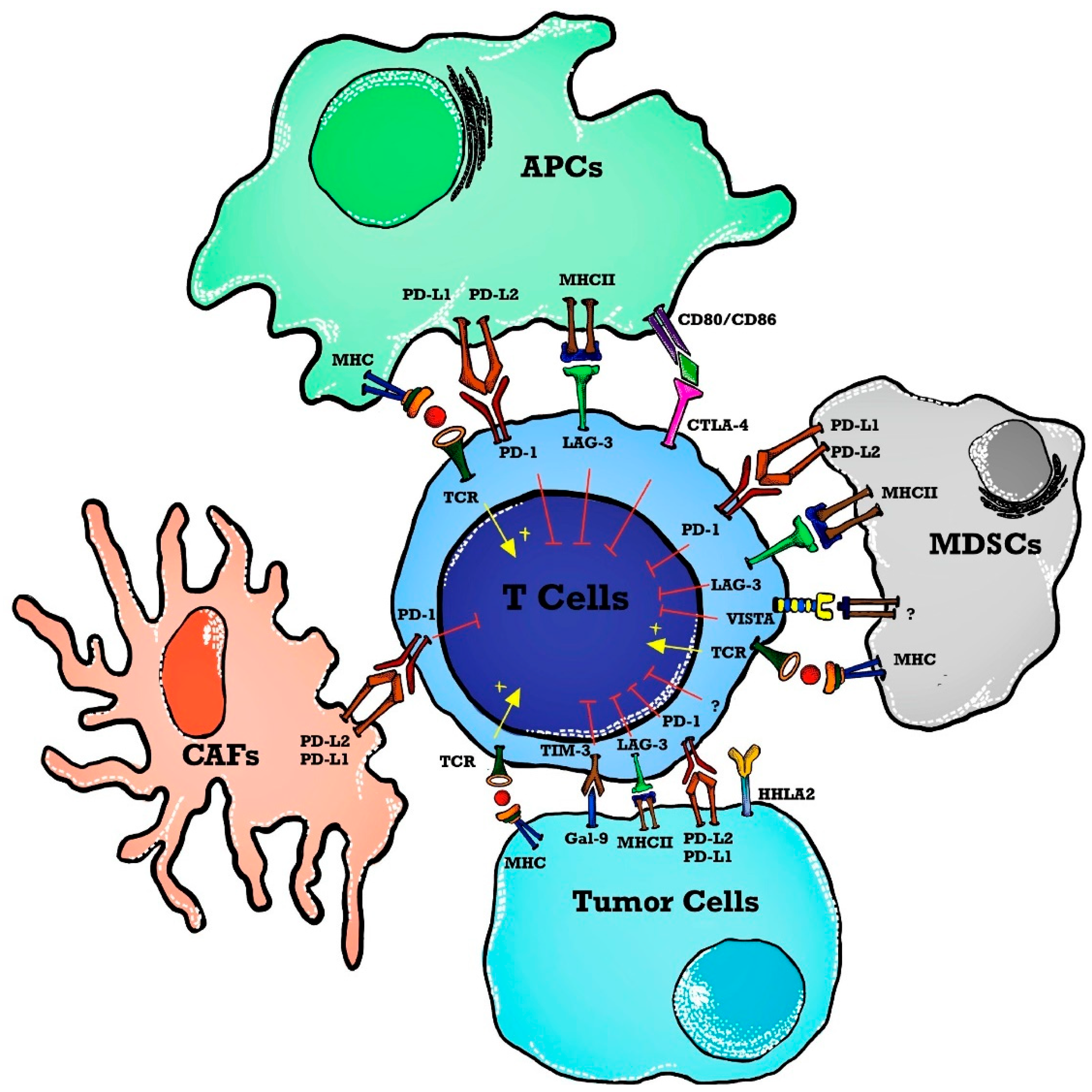
Figure 1. Immune checkpoints in a tumor microenvironment (TME). T cell activation is induced as APCs present tumor antigens to naïve T cells. The MHC and TCR signaling pathway provides a signal for T cell activation whereas immune checkpoints inhibit T cell activation in TME. Immune checkpoint markers are expressed on T cells, and ligands are present on APCs, tumor cells, and stromal cells such as CAFs and MDSCs. Abbreviations: APCs—antigen-presenting cells; MHC—major histocompatibility complex; TCR—T cell receptor; TME—tumor microenvironment; MDSCs—myeloid-derived suppressor cells; PD-1—programmed death 1; PD-L2—programmed cell death ligand-2; VISTA—V-domain immunoglobulin containing suppressor T cell activation; HHLA2—human endogenous retrovirus-H long terminal repeat-associated protein 2; TIM-3—T cell immunoglobulin and mucin domain 3; Gal-9—Galedctin-9; CAFs—cancer-associated fibroblasts; LAG-3—lymphocyte activated gene-3; CTLA-4—cytotoxic T-lymphocyte antigen-4.
2. LAG-3 Targeted Therapy and Its Role in Hematological Malignancies
Lymphocyte activation gene 3 (LAG-3) (CD223) is a CD4-related activation-induced cell surface inhibitory receptor that binds to major histocompatibility complex (MHC) class II molecules with high affinity and negatively regulates T cell effector functions [20]. Cells expressing LAG-3 are T cells, some activated B cells, plasmacytoid dendritic cells (DCs), and neurons [21]. LAG-3 ligands are MHC class-II, galectin-3 (Gal-3), and fibrinogen-like protein 1 (FGL1) with MHC-II being the canonical ligand [22]. LAG-3 binds to MHC class II with higher affinity than CD4 inducing rapid protein phosphorylation of phospholipase Cgamma2 (PLCgamma2) and p72syk as well as activation of phosphatidyl inositol 3-kinase/Akt, p42/44 extracellular signal-regulated protein kinase, and p38 mitogen-activated protein kinase pathways [23]. Gal-3 is expressed on tumor cells and activated T cells that are needed for CD8 T cell and plasmacytoid DC suppression [22]. FGL1 is highly produced by human cancer cells and binding of LAG-3 with FGL1 contributes to poor responses/resistance to anti-PD-1/anti-PD-L1 immunotherapies [24][25]. This mechanism forms the basis of PD-1 and LAG-3 co-blockade responsible for several T cell antitumor activities [26][27][28].
Currently, 16 LAG-3 targeted therapies are tested at 97 clinical trials by Bristol-Myers Squibb (BMS-986016), Regeneron Pharmaceuticals (REGN3767 and 89Zr-DFO-REGN3767), Merck (MK-4280), Novartis (LAG525), Tesaro (GSK) (TSR-033), Symphogen (Sym022), GlaxoSmith (GSK2831781), Incyte Biosciences International Sàrl (INCAGN02385), Prima BioMed/Immutep (IMP321), MacroGenics (MGD013), F-Star (FS118), Hoffmann-La Roche (RO7247669), Shanghai EpimAb Biotherapeutics (EMB-02), Xencor (XmAb841) and Innovent Biologics (IBI323) [29]. LAG-3 targeted therapies are divided into three categories namely monoclonal antibodies, soluble LAG-3—immunoglobulin (Ig) fusion proteins, and anti-LAG-3 bispecific drugs [29]. Most trials are Phase II (34), I/II (21), and II (35), and only two of them have reached Phase III for BMS-986016 (NCT05002569) [30] and MK-4280 drugs (NCT05064059) [31]. Table 1 demonstrates the use of LAG-3 agents in hematological malignancies in the current clinical trials.
Table 1. Summarizes all the clinical trials of LAG-3 therapy being used in hematological or related malignancies.
| NCT Number | Sponsors/Collaorators | Title | Drug Name | Phase | Tumor/Disease | Status | Outcome Measures | ICI Type | Study Designs |
|---|---|---|---|---|---|---|---|---|---|
| NCT04566978 | Memorial Sloan Kettering Cancer Center | 89Zr-DFO-REGN3767 in PET Scans in People With Diffuse Large B Cell Lymphoma (DLBCL) | Drug: 89Zr-DFO-REGN3767|Diagnostic Test: PET/CT | Early Phase 1 | Large B-cell Lymphoma|DLBCL | Recruiting | Biodistribution of 89Zr-DFO-REGN3767|Optimal 89Zr-DFO-REGN3767 mass dose for tumor targeting|Optimal time for imaging and tumor uptake post 89Zr-DFO-REGN3767 administration|Tumor lesion uptake of 89Zr-DFO-REGN3767 and correlate with LAG-3 expression by IHC | LAG-3 | Allocation: Randomized|Intervention Model: Sequential Assignment|Masking: None (Open Label)|Primary Purpose: Diagnostic |
| NCT03310619 | Celgene | A Safety and Efficacy Trial of JCAR017 Combinations in Subjects With Relapsed/Refractory B-cell Malignancies (PLATFORM) | Biological: JCAR017|Drug: Durvalumab|Drug: CC-122|Drug: Ibrutinib|Drug: CC-220|Drug: Relatlimab|Drug: Nivolumab|Drug: CC-99282 | Phase 1|Phase 2 | Lymphoma, Non-Hodgkin|Lymphoma, Large B-Cell, Diffuse|Lymphoma, Follicular | Completed | Dose-limiting toxicity (DLT) rates|Complete Response Rate|Adverse Events (AEs)|Progression-free survival (PFS)|Overall survival (OS)|Overall response rate (ORR)|Duration of response (DOR)|Event-free survival (EFS)|Pharmacokinetic (PK)-Cmax|Pharmacokinetic (PK)-Tmax|Pharmacokinetic (PK)-AUC|Health-related quality of life (HRQoL)|Quality of Life C30 questionnaire (EORTC-QLQ-C30)|European Quality of Life-5 Dimensions health state classifier to 5 Levels (EQ-5D-5L) | LAG-3 | Allocation: Randomized|Intervention Model: Parallel Assignment|Masking: None (Open Label)|Primary Purpose: Treatment |
| NCT02061761 | Bristol-Myers Squibb | A Phase 1/2a Dose Escalation and Cohort Expansion Study of the Safety, Tolerability, and Efficacy of Anti-LAG-3 Monoclonal Antibody (Relatlimab, BMS-986016) Administered Alone and in Combination With Anti-PD-1 Monoclonal Antibody (Nivolumab, BMS-936558) in Relapsed or Refractory B-Cell Malignancies | Biological: BMS-986016|Biological: BMS-936558 | Phase 1|Phase 2 | Hematologic Neoplasms | Completed | Safety measured by the rate of Adverse events (AEs), Serious Adverse events (SAEs), death and laboratory abnormalities [Time Frame: Up to approximately 2.3 years] | LAG-3 | Allocation: Non-Randomized|Intervention Model: Single Group Assignment|Masking: None (Open Label)|Primary Purpose: Treatment |
| NCT03598608 | Merck Sharp & Dohme LLC | A Phase 1/Phase 2 Clinical Study to Evaluate the Safety and Efficacy of a Combination of MK-4280 and Pembrolizumab (MK-3475) in Participants With Hematologic Malignancies | Biological: pembrolizumab|Biological: Favezelimab | Phase 1|Phase 2 | Hodgkin Disease|Lymphoma, Non-Hodgkin|Lymphoma, B-Cell | Recruiting | Percentage of Participants Experiencing a Dose-limiting Toxicity (DLT); Percentage of Participants Experiencing an Adverse Event (AE); Percentage of Participants with Treatment Discontinuations Due to an AE | LAG-3 | Allocation: Non-Randomized|Intervention Model: Parallel Assignment|Masking: None (Open Label)|Primary Purpose: Treatment |
| NCT04913922 | Ludwig-Maximilians-University of Munich | An Open-Label Phase II Study of Relatlimab (BMS-986016) With Nivolumab (BMS-936558) in Combination With 5-Azacytidine for the Treatment of Patients With Refractory/Relapsed Acute Myeloid Leukemia and Newly Diagnosed Older Acute Myeloid Leukemia Patients | Drug: Azacitidine Injection|Drug: Nivolumab|Drug: Relatlimab | Phase 2 | Acute Myeloid Leukemia | Recruiting | Maximum tolerated dose (MTD); Dose-limiting toxicities (DLTs); Objective response rate (ORR) | LAG-3 | Allocation: N/A|Intervention Model: Single Group Assignment|Masking: None (Open Label)|Primary Purpose: Treatment |
| NCT03365791 | Novartis Pharmaceuticals | Modular Phase 2 Study to Link Combination Immune-therapy to Patients With Advanced Solid and Hematologic Malignancies. Module 9: PDR001 Plus LAG525 for Patients With Advanced Solid and Hematologic Malignancies. | Biological: PDR001; Biological: LAG525 | Phase 2 | Small cell lung cancer, Gastric/esophageal adenocarcinoma, Castration resistant prostate adenocarcinoma (CRPC), Soft tissue sarcoma, Ovarian adenocarcinoma, Advanced well-differentiated neuroendocrine tumors, Diffuse large B cell lymphoma (DLBCL). | Completed | Clinical Benefit Rate (CBR) at 24 Weeks of PDR001+LAG525 by Tumor Type in Multiple Solid Tumors and Lymphoma | LAG-3 | Allocation: N/A|Intervention Model: Single Group Assignment|Masking: None (Open Label)|Primary Purpose: Treatment |
-
Use of 89Zr-DFO-REGN3767 in PET scans of people with diffuse large B cell lymphomas (DLBCL) was the pilot study (NCT04566978) [32] undertaken at Memorial Sloan Kettering Hospital in 2022 with the main purpose of the study looking at the way the body absorbs, distributes, and gets rid of 89Zr-DFO-REGN3767 [33]. 89Zr-DFO-REGN3767 is comprised of the anti-LAG-3 antibody, REGN3767 labeled with the positron-emitter zirconium-89 (89Zr) through the chelator-linker DFO and REGN3767 is an investigational monoclonal antibody that targets LAG-3 receptors. This study is a diagnostic research study determining the optimal time for imaging and tumor uptake post 89Zr-DFO-REGN3767 administration. However, it can help evaluate tumor lesion uptake of 89Zr-DFO-REGN3767 and correlate with LAG-3 expression by immunohistochemistry (IHC) in tumors that will be compared descriptively with other biomarkers of TME characterized in biopsies, such as quantitation of IHC score (LAG-3 and/or other immune cell markers).
-
A safety and efficacy trial of JCAR017 (lisocabtagene maraleucel, also known as liso-cel) (a CD19-targeted chimeric antigen receptor CART-cell therapy) combinations in subjects with relapsed/refractory B cell malignancies (PLATFORM) (NCT03310619) [34] was performed. Relatlimab, BMS-986016 is an anti-LAG-3 fully human monoclonal IgG4-κ antibody that binds human LAG-3 with high affinity and inhibits its binding to MHC-II [35]. This trial was a global, open-label, multi-arm, parallel multi-cohort, multi-center, Phase I/II study to determine the safety, tolerability, pharmacokinetics, efficacy, and patient-reported quality of life of JCAR017 in combination with various agents including relatlimab, durvalumab, avadomide, iberdomide, ibrutinib, and nivolumab. The trial was completed, and the studied tumors were non-Hodgkin lymphoma (NHL), diffuse large B cell lymphoma (DLBCL), and Follicular lymphoma (FL). The objective of this study was that during Phase I, different arms may be opened to test JCAR017 in combination with combination agent(s) in adult subjects with R/R aggressive B cell NHL. Within each arm, different doses and schedules of JCAR017 and the combination agent(s) may be tested in several cohorts and subcohorts per arm. During Phase II of this study, the expansion of any dose level and schedule for any arm that is safe may occur. All subjects from Phase I and Phase II will be followed for 24 months for survival, relapse, long-term toxicity, and viral vector safety as per guidelines.
-
A similar trial was also designed with relatlimab by Bristol-Myers Squibb, NCT02061761 [36] administered alone or in combination with nivolumab to subjects with relapsed or refractory B cell malignancies (relapsed or refractory Hodgkin lymphoma (HL) and relapsed or refractory DLBCL and to study its safety, tolerability, dose-limiting toxicities and maximum tolerated dose. The trial completed and studied hematological malignancies including chronic lymphocytic leukemia (CLL), HL, NHL, and Multiple Myeloma (MM). A detailed description of dose-related adverse events was studied and was +displayed in the result section of the trial.
-
Favezelimab (MK-4280) is another LAG-3 antibody that is studied in combination with pembrolizumab (MK-3475) in the clinical trial NCT03598608 [37] that was started in July 2018 to study and evaluate the safety and efficacy of these agents in hematologic malignancies. It included classical HL, DLBCL, and indolent HL. No results have been posted till the writing of this research. This study will also evaluate the safety and efficacy of pembrolizumab or favezelimab administered as monotherapy in participants with classical HL using a 1:1 randomized study design.
-
Relapsed or refractory acute myeloid leukemia (AML) and newly diagnosed older AML are included in the ClinicalTrials.gov Identifier: NCT04913922 [38] to study the combination of relatlimab with nivolumab and 5-azacytidine. No results have been posted yet.
All the above trials included LAG-3 as an ICI agent in the above-mentioned hematological malignancies, however, no trials have been conducted in the field of MPN.
3. Mechanism of Action of LAG-3
In 1990, Triebel and colleagues discovered LAG-3, a novel type I transmembrane protein consisting of 498 amino acids, which is expressed on activated human natural killer (NK) and T cell lines [39]. The LAG-3 gene is located in close proximity to CD4 on chromosome 12 in humans (chromosome 6 in mice). Structurally, LAG-3 exhibits high similarity to CD4, featuring four extracellular immunoglobulin superfamily (IgSF)-like domains (D1-D4) [40]. These structural motifs are conserved between LAG-3 and CD4, resulting in similar extracellular folding patterns. Consequently, LAG-3 can bind to major histocompatibility complex (MHC) class II molecules, albeit at a distinct site, with even greater affinity than CD4 [41]. LAG-3 was suggested to be spatially associated with the T cell receptor TCR: CD3 complex present in lipid raft microdomains to allow for the clustering of signaling molecules and the formation of the immunological synapse. However, the exact mechanism is still unclear [42]. LAG-3 lacks a binding site in the cytoplasmic tail for the tyrosine kinase p56Lck, which CD4 uses to promote signal transduction downstream of the T cell receptor (TCR) [41]. Instead, the LAG-3 cytoplasmic domain appears to have three well-defined motifs namely a serine-based motif that could act as a PKC substrate, a repetitive “EP” motif consisting of a series of glutamic acid-proline dipeptide repeats, and a relatively unique “KIEELE” motif, highlighted by an essential lysine residue [43][44]. The absence of the cytoplasmic tail in LAG3 mutants reveals an intriguing aspect of its function, as these mutants neither compete with CD4 nor mediate the inhibitory effects typically associated with LAG3 [20]. This observation highlights the importance of transmitting an inhibitory signal through LAG3’s cytoplasmic domain. Notably, the expression of MHC class II molecules on human melanoma cells is linked to unfavorable prognoses. In the context of melanoma-infiltrating T cells, the high expression of LAG3 and its interaction with MHC class II molecules may contribute to clonal exhaustion [45]. This interaction, demonstrated in vitro, could potentially serve as an evasion mechanism employed by tumor cells, safeguarding them against apoptosis. Recent studies indicate that melanoma cells expressing MHC class II molecules attract a specific infiltration of CD4+ T cells, potentially facilitated by the interaction between LAG3 and MHC class II molecules. Consequently, this interaction negatively impacts CD8+ T cell responses [46][47]. These findings shed light on the intricate interplay between LAG3, MHC class II molecules, and tumor cells, ultimately influencing immune responses and highlighting LAG3 as a key modulator in cancer immunology. Galectin-3, a ligand expressed by numerous cells within the tumor microenvironment rather than the tumor itself, has the potential to interact with LAG3 on tumor-specific CD8+ T cells, thereby modulating anti-tumor immune responses [48]. Another interesting molecule, liver sinusoidal endothelial cell lectin (LSECtin), is present in the liver and has also been detected in human melanoma tissues, where it facilitates tumor growth by inhibiting T cell-dependent anti-tumor responses [49]. Interaction between LAG-3 and LSECtin in melanoma cells has been found to impede IFNγ production by antigen-specific effector T cells, thereby altering the tumor microenvironment [49]. The persistence of T cell activation within a chronic inflammatory environment, particularly in the presence of tumors, often leads to the sustained co-expression of LAG3 and other inhibitory receptors (IR) such as PD1, TIGIT, TIM3, CD160, and 2B4, resulting in a state of T cell dysfunction [50]. Although LAG3 is widely expressed across various hematopoietic cell types, including CD11clow B220+ PDCA-1+ plasmacytoid dendritic cells (pDCs) which exhibit higher levels of LAG3 expression compared to other subsets, it is not expressed on any myeloid or lymphoid DC subset [51]. In vitro experiments have demonstrated that MHC class II-expressing melanoma cells can stimulate LAG3+ pDCs to mature and produce IL-6, a finding confirmed in vivo where LAG3+ pDCs displayed elevated IL-6 production and an activated phenotype in close proximity to melanoma cells [52]. Furthermore, a study by Bo Huang et al. suggests that increased IL-6 levels prompt the release of CCL2 by monocytes in vitro, which in turn may recruit MDSCs, thus proposing the hypothesis that LAG3+ pDCs may indirectly drive MDSC-mediated immunosuppression through engagement with MHC class II+ melanoma cells [53]. The activity of LAG-3 is also regulated through cell surface cleavage mediated by ADAM10 and ADAM17 disintegrin/metalloproteases. However, it is important to note that soluble LAG-3 does not seem to possess any biological function in mice [54].
References
- Passamonti, F.; Mora, B.; Maffioli, M. New molecular genetics in the diagnosis and treatment of myeloproliferative neoplasms. Curr. Opin. Hematol. 2016, 23, 137–143.
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228.
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719.
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679.
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398.
- JJames, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061.
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397.
- Barosi, G. An Immune Dysregulation in MPN. Curr. Hematol. Malign-Rep. 2014, 9, 331–339.
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392.
- Zhao, W.B.; Li, Y.; Liu, X.; Zhang, L.Y.; Wang, X. Involvement of CD4+CD25+ regulatory T cells in the pathogenesis of polycythaemia vera. Chin. Med. J. 2008, 121, 1781–1786.
- Skov, V.; Riley, C.H.; Thomassen, M.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leuk. Lymphoma 2013, 54, 2269–2273.
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195.
- Bjørn, M.E.; Hasselbalch, H.C. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, e648090.
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; Sindhu, H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk. Res. 2016, 43, 39–43.
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857.
- Dalle, I.A.; Kantarjian, H.; Daver, N.; Masarova, L.; Pemmaraju, N.; Bose, P.; Garcia-Manero, G.; Verstovsek, S. Phase II study of single-agent nivolumab in patients with myelofibrosis. Ann. Hematol. 2021, 100, 2957–2960.
- Hobbs, G.; Bozkus, C.C.; Moshier, E.L.; Dougherty, M.; Bar-Natan, M.; Sandy, L.; Johnson, K.; Foster, J.E.; Som, T.; Macrae, M.; et al. PD-1 inhibition in advanced myeloproliferative neoplasms. Blood Adv. 2021, 5, 5086–5097.
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes. Cancer 2018, 9, 176–189.
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting Edge: Molecular Analysis of the Negative Regulatory Function of Lymphocyte Activation Gene-3. J. Immunol. 2002, 169, 5392–5395.
- Saleh, R.; Toor, S.M.; Nair, V.S.; Elkord, E. Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front. Immunol. 2020, 11, 1469.
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423.
- Andreae, S.; Buisson, S.; Triebel, F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood 2003, 102, 2130–2137.
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.
- Zuazo, M.; Arasanz, H.; Fernández-Hinojal, G.; García-Granda, M.J.; Gato, M.; Bocanegra, A.; Martínez, M.; Hernández, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD 4 immunity is required for clinical responses to PD-L1/ PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293.
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 2018, 9, 385.
- Jing, W.; Gershan, J.A.; Weber, J.; Tlomak, D.; McOlash, L.; Sabatos-Peyton, C.; Johnson, B.D. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J. Immunother. Cancer 2015, 3, 2.
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880.
- Chocarro, L.; Blanco, E.; Arasanz, H.; Fernández-Rubio, L.; Bocanegra, A.; Echaide, M.; Garnica, M.; Ramos, P.; Fernández-Hinojal, G.; Vera, R.; et al. Clinical landscape of LAG-3-targeted therapy. Immuno-Oncol. Technol. 2022, 14, 100079.
- Bristol-Myers Squibb. A Phase 3, Randomized, Double-Blind Study of Adjuvant Immunotherapy with Nivolumab + Relatlimab Fixed-Dose Combination Versus Nivolumab Monotherapy after Complete Resection of Stage III–IV Melanoma. clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05002569 (accessed on 9 March 2023).
- Merck Sharp & Dohme LLC. A Phase 3 Study of MK-4280A (Coformulated Favezelimab Plus Pembrolizumab ) Versus Standard of Care in Previously Treated Metastatic PD-L1 Positive Colorectal Cancer. clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05064059 (accessed on 9 March 2023).
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34.
- Memorial Sloan Kettering Cancer Center. A Pilot Study of 89Zr-DFO-REGN3767 Anti LAG-3 Antibody Positron Emission Tomography in Patients with Relapsed/Refractory DLBCL. clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04566978 (accessed on 12 March 2023).
- Celgene. An Exploratory Phase 1/2 Trial To Evaluate The Safety And Efficacy Of JCAR017 Combinations In Subjects with Relapsed/Refractory B-Cell Malignancies (PLATFORM). clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT03310619 (accessed on 12 March 2023).
- Albershardt, T.C.; Parsons, A.J.; Reeves, R.S.; Flynn, P.; Campbell, D.J.; ter Meulen, J.; Berglund, P. Therapeutic efficacy of PD1/PDL1 blockade in B16 melanoma is greatly enhanced by immunization with dendritic cell-targeting lentiviral vector and protein vaccine. Vaccine 2020, 38, 3369–3377.
- Bristol-Myers Squibb. A Phase 1/2a Dose Escalation and Cohort Expansion Study of the Safety, Tolerability, and Efficacy of Anti-LAG-3 Monoclonal Antibody (Relatlimab, BMS-986016) Administered Alone and in Combination with Anti-PD-1 Monoclonal Antibody (Nivolumab, BMS-936558) in Relapsed or Refractory B-Cell Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02061761 (accessed on 12 March 2023).
- Merck Sharp & Dohme LLC. A Phase 1/Phase 2 Clinical Study to Evaluate the Safety and Efficacy of a Combination of MK-4280 and Pembrolizumab (MK-3475) in Participants with Hematologic Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT03598608 (accessed on 13 March 2023).
- MD VB. An Open-Label Phase II Study of Relatlimab (BMS-986016) with Nivolumab (BMS-936558) in Combination with 5-Azacytidine for the Treatment of Patients with Refractory/Relapsed Acute Myeloid Leukemia and Newly Diagnosed Older Acute Myeloid Leukemia Patients. clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04913922 (accessed on 13 March 2023).
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Wang, J.-H.; Meijers, R.; Xiong, Y.; Liu, J.-H.; Sakihama, T.; Zhang, R.; Joachimiak, A.; Reinherz, E.L. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc. Natl. Acad. Sci. USA 2001, 98, 10799–10804.
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749.
- Hannier, S.; Triebel, F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. Int. Immunol. 1999, 11, 1745–1752.
- Mastrangeli, R.; Micangeli, E.; Donini, S. Cloning of Murine LAG-3 by Magnetic Bead Bound Homologous Probes and PCR (GENE-CAPTURE PCR). Anal. Biochem. 1996, 241, 93–102.
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96.
- Ruiter, D.J.; Mattijssen, V.; Broecker, E.B.; Ferrone, S. MHC antigens in human melanomas. Semin. Cancer Biol. 1991, 2, 35–45.
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC Class II Engagement by Its Ligand LAG-3 (CD223) Contributes to Melanoma Resistance to Apoptosis. J. Immunol. 2011, 186, 5173–5183.
- Donia, M.; Andersen, R.; Kjeldsen, J.W.; Fagone, P.; Munir, S.; Nicoletti, F.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+ T-Cells, Which Dampen CD8+ T-cell Antitumor Reactivity. Cancer Res. 2015, 75, 3747–3759.
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635.
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-cell Responses. Cancer Res. 2014, 74, 3418–3428.
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864.
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A.A. LAG-3 Regulates Plasmacytoid Dendritic Cell Homeostasis. J. Immunol. 2009, 182, 1885–1891.
- Camisaschi, C.; De Filippo, A.; Beretta, V.; Vergani, B.; Villa, A.; Vergani, E.; Santinami, M.; Cabras, A.D.; Arienti, F.; Triebel, F.; et al. Alternative Activation of Human Plasmacytoid DCs In Vitro and in Melanoma Lesions: Involvement of LAG-3. J. Investig. Dermatol. 2014, 134, 1893–1902.
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.-M.; et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92.
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
943
Revisions:
2 times
(View History)
Update Date:
21 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No