Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmed Elshazly | -- | 1897 | 2023-08-10 13:15:23 | | | |
| 2 | Catherine Yang | Meta information modification | 1897 | 2023-08-11 02:56:57 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Elshazly, A.M.; Sinanian, M.M.; Elimam, D.M.; Zakaria, S. β-Amyloid and Tau Protein in Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/47904 (accessed on 27 July 2026).
Elshazly AM, Sinanian MM, Elimam DM, Zakaria S. β-Amyloid and Tau Protein in Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/47904. Accessed July 27, 2026.
Elshazly, Ahmed M., Melanie M. Sinanian, Diaaeldin M. Elimam, Sherin Zakaria. "β-Amyloid and Tau Protein in Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/47904 (accessed July 27, 2026).
Elshazly, A.M., Sinanian, M.M., Elimam, D.M., & Zakaria, S. (2023, August 10). β-Amyloid and Tau Protein in Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/47904
Elshazly, Ahmed M., et al. "β-Amyloid and Tau Protein in Alzheimer’s Disease." Encyclopedia. Web. 10 August, 2023.
Copy Citation
Alzheimer’s disease (AD) is one of the major causes of dementia and its incidence represents approximately 60–70% of all dementia cases worldwide. Many theories have been proposed to describe the pathological events in AD, including deterioration in cognitive function, accumulation of β-amyloid, and tau protein hyperphosphorylation. Infection as well as various cellular molecules, such as apolipoprotein, micro-RNA, calcium, ghrelin receptor, and probiotics, are associated with the disruption of β-amyloid and tau protein hemostasis.
Alzheimer’s
β-amyloid
tau protein
calcium
probiotics
1. Introduction
Alzheimer’s disease (AD) is the most frequent cause of dementia, accounting for 60–70% of all cases globally [1][2]. In 2019, AD was ranked the sixth most common cause of death in the US, while in 2020 and 2021 AD was the seventh leading cause of death in the US [3]. Furthermore, AD is the fifth most common cause of death in US citizens aged 65 and older [4]. Various molecular theories have been proposed to explain the pathological events in AD, including the accumulation of β-amyloid and hyperphosphorylation and aggregation of tau protein. The disruption of β-amyloid and tau protein hemostasis are associated with several cellular molecules and interactions, such as apolipoprotein, micro-RNA, calcium, and ghrelin receptor (GHSR1α). Gut microflora and infection are also associated with their dysregulation. This research will shed a light on the integrating cellular and signaling molecules that may have a complementary role in β-amyloid and tau protein dysregulation (Figure 1).
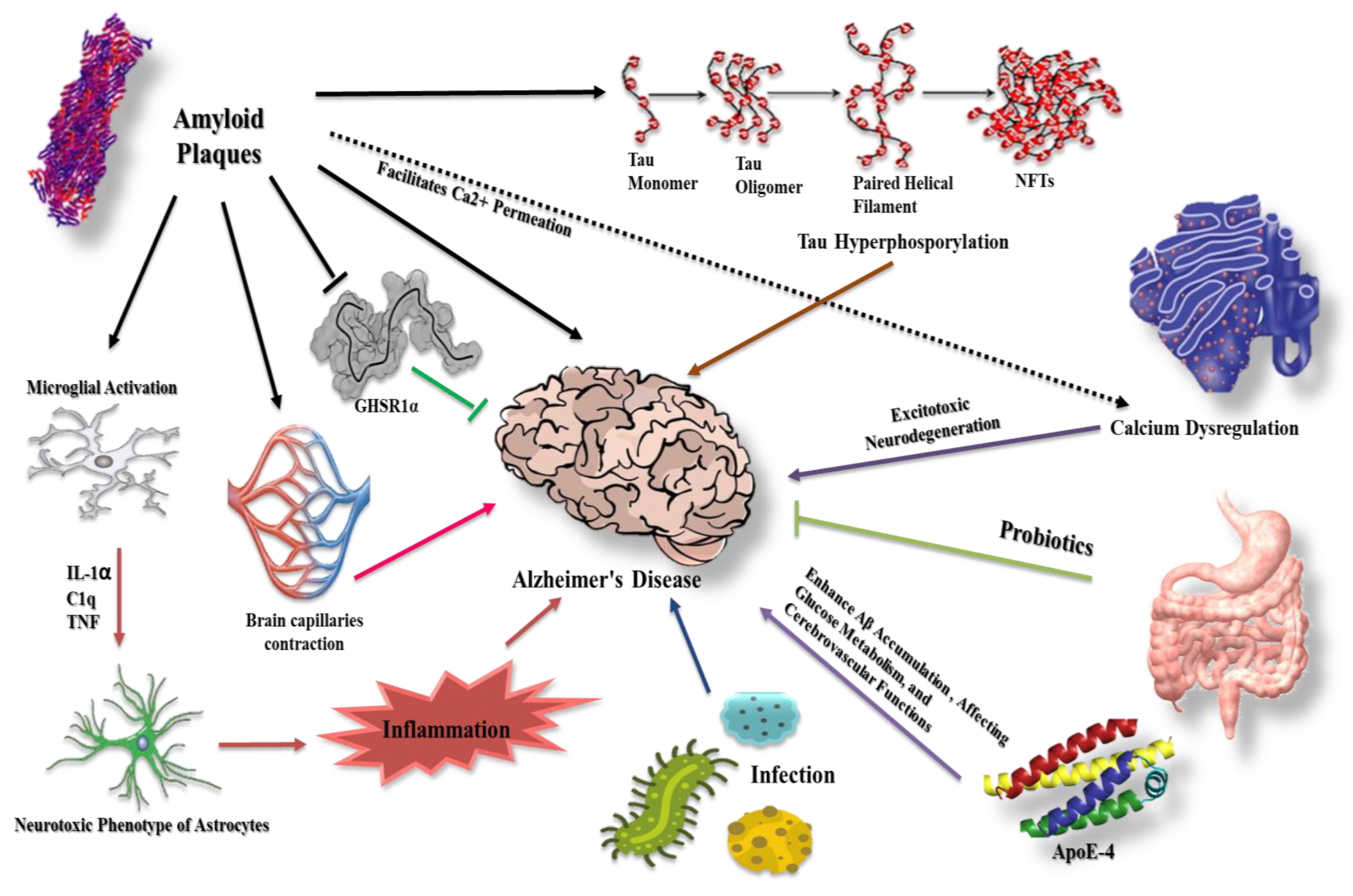
Figure 1. Different molecular modalities and signaling pathways intersecting with β-amyloid and tau protein in AD. β-amyloid plaques affect several pathways that may contribute to AD progression. For example, they facilitate Ca2+ permeation, eventually leading to neurotoxicity, and increase the extent of inflammation via microglial and astrocyte activation. Furthermore, β-amyloid counteracts the protective effect of GHSR1α and causes cerebral capillary vasocontraction while potentiating tau phosphorylation. Tau hyperphosphorylation has a role in AD etiology together with infection and apolipoprotein E-4 (ApoE-4). Probiotics demonstrate protective effects.
2. β-Amyloid in Alzheimer’s Disease
β-amyloid (Aβ) aggregation has long been thought to be a key and first event in the etiology of Alzheimer’s disease. Amyloid protein precursors (APPs) are enzymatically cleaved into Aβ peptides that range in length from 38 to 43 amino acids [5], giving rise to oligomers, polymers, and eventually insoluble amyloid aggregates upon linking with each other [6]. Aβ released by neurons, then enters the bloodstream and cerebrospinal fluid (CSF), with clearance mechanisms preventing Aβ deposition physiologically. However, the imbalance between Aβ production and clearance can result in its sedimentation [6], causing synaptic and neuronal dysregulation [7][8], and eventually contributing to AD pathogenesis [9][10]. Furthermore, the relationship between apoptosis and Aβ have been investigated, for example, Yao et al. [11] showed that Aβ affect the expression of Bcl2 family proteins. Aβ significantly reduce expression of the antiapoptotic proteins, Bcl-w and Bcl-xL, together with increasing the mitochondrial release of second mitochondrion-derived activator of caspase (Smac) [11]. Han et al. [12] reported that Aβ induces apoptosis via promoting mitochondrial fission, disrupting mitochondrial membrane potential, increasing intracellular reactive oxygen species (ROS) level, and activating the process of mitophagy. Barrantes et al. [13] also showed that Aβ 42 causes DNA strand breaks, leading to p53-mediated apoptosis.
3. Tau Protein and Formation of Neurofibrillary Tangles
Neurofibrillary pathology is one of the most common characteristics of AD, and includes neurotic plaques, neurofibrillary tangles, and threads generated by tau protein aggregation, which is a microtubule-associated protein [6]. In pathological situations, tau undergoes hyperphosphorylation, forming insoluble fibers (“paired helical filaments”) [14]. Hyperphosphorylated paired helical filaments combine to generate neurofibrillary tangles (NFTs).
Physiologically, tau phosphorylation is controlled by the balance between kinases and phosphatases [14][15]. This balance can be disrupted via oxidative stress and through an increase in the activity of protein kinases, mainly glycogen synthase kinase 3 (GSK-3𝛽), which has been shown to be upregulated in AD [15]. Furthermore, the reduced phosphatase activity in the brain of AD patients can augment the hyperphosphorylation induced by protein kinases [16][17].
Lovell et al. [18] has shown that tau phosphorylation is significantly increased with higher GSK-3𝛽 activity in primary rat cortical neuron cultures, stimulated through cuprizone, a copper chelator, in combination with oxidative stress (Fe2+/H2O2) [18]. Moreover, they demonstrated a reduction in tau hyperphosphorylation through the inhibition of GSK-3𝛽 activity with lithium, as confirmed by transglutaminase 3 staining [18]. Consistently with Lovell et al., Su et al. [19] showed that a fragment of tau protein possesses copper reduction activity, initiating the copper-mediated generation of hydrogen peroxide. The generated hydrogen peroxide has been shown to increase GSK-3𝛽 activity, causing tau hyperphosphorylation in human embryonic renal cells 293 [17].
Tau hyperphosphorylation also affects the stability of microtubules, resulting in axonal and neural dysfunction [20]. Therefore, many efforts have been made to utilize microtubule-stabilizing drugs in AD. For example, Zhang et al. [21] showed that the administration of paclitaxel in mouse models with tau pathology restored fast axonal transport in spinal axons and improved motor impairment. Recently, Zhang et al. [22] showed that triazolopyrimidine, a microtubule-stabilizing drug, significantly lowered tau pathology and improved cognitive function in transgenic mouse models of tauopathy.
4. Prion-like Conformation of β-Amyloid and Tau Proteins
Prion protein (PrPSc) is a unique protein form that has enhanced infectivity, self-replication ability, and persistent survival—even in the denaturing conditions of the gut. In humans, prions can cause different neuronal disorders, including Creutzfeldt–Jakob disease (CJD), and fatal familial insomnia (FFI) [23], which can either develop spontaneously from hereditary factors or as a result of infection.
Aβ aggregates into different forms, including polymorphic amyloid fibrils and a variety of intermediate assemblies, including oligomers and protofibrils [24]. Several studies reported that Aβ spreads through the brain in a harmful configuration similar to PrPSc [25]. Condello et al. demonstrated that the injection of brain-derived Aβ from AD patients into the brains of transgenic mice exhibited a prion-like appearance [26] together with significant Aβ deposition. In addition, in the human brain prion-like Aβ forms in one or more regions before spreading to other regions, suggesting cross-synaptic transmission [27]. Pignataro et al. [28] discussed different findings to explain how Aβ spreads through the brain regions. One hypothesis mentioned by Pignataro et al. [28] is that Aβ can spread in the AD brain by advancing through synaptically connected regions since Aβ is released from synaptic terminals, thus making brain nodes vulnerable to Aβ accumulation.
Tau protein has been reported to spread in a prion-like manner in the brain, although earlier investigations focused mainly on total insoluble tau, as the presence of NFT correlates with the degree of brain atrophy and cognitive impairment in AD [29]. Clifford et al. reported that low activity of prion-like tau has been linked to extended life spans. Furthermore, they showed that both prion-like Aβ and prion-like tau proteins were found in 100 postmortem brain tissue samples from patients who died of AD [30]. Levels of both prion-like Aβ and prion-like tau are reported to be associated with age and dysregulation in production or clearance [27], however, more investigation is still required to uncover the mechanisms by which prion-like Aβ and prion-like tau proteins spread through the brain regions, in addition to the ability to target these conformations.
5. The Intervention of Glial Cells in β-Amyloid and Tau Protein Dysregulation
5.1. Astrocytes
Various neuronal cells, including astrocytes and microglia, help in maintaining the physiological levels of Aβ and tau protein. Astrocytes are specialized glial cells that construct the central nervous system (CNS) scaffold and can be found in two forms: fibrous (mostly in white matter) and protoplasmic (mostly in gray matter) [31]. Astrocytes contribute to various physiological activities, including fluid maintenance, cerebral blood flow regulation, neurotransmitter balance, and synaptic hemostasis [32]. Astrocytes also constitute the glymphatic system, which eliminates neurotoxic waste products such as amyloid and tau species [33]. Astrocytes, which are near amyloid plaques in human brains, have been found to contain amyloid-containing granules, implying that astrocytes work to remove amyloid accumulation during the disease progression [34]. In vitro and in vivo investigations have revealed that astrocytes move towards amyloid plaques and work to clear Aβ aggregation [35].
Astrocytes have two major phenotypes: A1, which is neurotoxic, and A2, which is thought to be protective. A1 astrocytes are expressed by activation of inflammatory cascades, mainly through NF-κB induction, a finding that is supported by the abundance of A1 astrocytes in postmortem brain tissues from persons with AD [36]. On the other hand, the A2 phenotype is induced by ischemia through activation of signal transducers and activators of the transcription 3 (STAT3) pathway. The neurotoxic A1 cells are marked by the expression of inflammatory mediators, while the protective A2 cells are marked by the expression of neurotrophic factors [37].
Interestingly, in animal models of AD, reactive astrocytes were discovered to release excessive GABA and glutamate, leading to impaired memory and synaptic loss [38]. Moreover, these cells contributed to microcirculation dysregulation and disruption of the blood–brain barrier (BBB), which facilitated Aβ accumulation and therefore disease progression [39]. Different molecules have been studied as possible treatments of AD through modulation of astrocyte phenotypes. One of these molecules is minocycline, where its intrathecal injection drastically downregulated A1 and upregulated A2 astrocyte levels [40][41].
5.2. Microglia
Microglia are innate immune cells of the myeloid lineage that exist in the CNS. Microglial activity is thought to be involved in CNS development, maturation, and senescence via the modulation of different regulatory networks [42]. Microglia play critical roles in neuronal apoptosis, synaptic maintenance, immune surveillance, and developmental synaptic pruning, in which the process of removing embryonic excess synapses improves the effectiveness of the neural network [42][43]. Dysregulated synaptic pruning is thought to be linked to autism disorders; moreover, it may be associated with weakened immune surveillance functions found in various neurodegenerative diseases [44][45].
Microglia express pattern recognition receptors (PRRs) recognizing two types of ligands: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs, including Aβ species). These receptors are responsible for triggering a microglial response in the presence of an exogenous or endogenous pathological insult [46]. The pathogenic species are then internalized by activated microglia via pinocytosis, phagocytosis, or receptor-mediated endocytosis. Through such endocytic processes, microglia attempt to degrade such pathogens and insults, additionally activating the release of various molecules, including interferons and chemokine receptors [47]. This process generally ceases once the immunological stimulus is removed, however, microglia have functional impairments and may be persistently activated in older brains, playing a role in AD pathogenesis [48].
Under pathological conditions, the morphology of microglia changes depending on the stage of the disease. In mouse models of AD, transcriptomic investigations have revealed that disease development is mirrored in microglia by progressive changes from a homeostatic to a disease-associated state, including less branching, which limits their surveillance functions [49]. The transition to the latter stage is often associated with the downregulation of homeostatic genes and upregulation of AD-associated genes, including apolipoprotein E (ApoE), protein tyrosine kinase binding protein (TYROBP), and triggering receptor expressed on myeloid cells 2 (TREM2) [50]. TREM2 plays a critical role in microglial activation processes, suggesting its possible role in the pathogenesis of neuronal disorders [51]. These studies confirmed the effects of aging on the human microglial phenotypes, including the downregulation of genes encoding cytoskeleton proteins, adhesion molecules, and cell surface receptors as well as the upregulation of certain genes, such as chemokine receptor type 4 (CXCR4), vascular endothelial growth factor 4 (VEGF4), and interleukin-15 (IL-15) [52].
5.3. Cellular Cross Talk between Astrocyte and Microglia
Aβ has been found to stimulate the NF-κB pathway in astrocytes, increasing complement C3 release, which then causes neuronal impairment and microglial activation [53]. The activated microglia secrete several factors, including interleukin 1 alpha (IL-1α), tumor necrosis factor (TNF), and complement component 1q (C1q), inducing astrocyte differentiation to the A1 phenotype [36]. Under AD inflammatory conditions, the interplay between astrocytes and microglia may generate a positive feedback loop, causing an inflammatory response [54].
References
- Hussein, W.; Sağlık, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules 2018, 23, 2033.
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 2000, 54, 2072–2077.
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2023, 19, 1598–1695.
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2021, 17, 327–406.
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619.
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113.
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112.
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235.
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185.
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1149–1158.
- Han, X.J.; Hu, Y.Y.; Yang, Z.J.; Jiang, L.P.; Shi, S.L.; Li, Y.R.; Guo, M.Y.; Wu, H.L.; Wan, Y.Y. Amyloid β-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 2017, 16, 4521–4528.
- Barrantes, A.; Rejas, M.T.; Benítez, M.J.; Jiménez, J.S. Interaction between Alzheimer’s Abeta1-42 peptide and DNA detected by surface plasmon resonance. J. Alzheimer’s Dis. 2007, 12, 345–355.
- Barghorn, S.; Davies, P.; Mandelkow, E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 2004, 43, 1694–1703.
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128.
- Feng, Y.; Xia, Y.; Yu, G.; Shu, X.; Ge, H.; Zeng, K.; Wang, J.; Wang, X. Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H2O2. J. Neurochem. 2013, 126, 234–242.
- Lovell, M.A.; Xiong, S.; Xie, C.; Davies, P.; Markesbery, W.R. Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimer’s Dis. 2004, 6, 659–671.
- Su, X.Y.; Wu, W.H.; Huang, Z.P.; Hu, J.; Lei, P.; Yu, C.H.; Zhao, Y.F.; Li, Y.M. Hydrogen peroxide can be generated by tau in the presence of Cu(II). Biochem. Biophys. Res. Commun. 2007, 358, 661–665.
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222.
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231.
- Zhang, B.; Yao, Y.; Cornec, A.S.; Oukoloff, K.; James, M.J.; Koivula, P.; Trojanowski, J.Q.; Smith, A.B., 3rd; Lee, V.M.; Ballatore, C.; et al. A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model. Mol. Neurodegener. 2018, 13, 59.
- Adaikkan, C.; Middleton, S.J.; Marco, A.; Pao, P.C.; Mathys, H.; Kim, D.N.; Gao, F.; Young, J.Z.; Suk, H.J.; Boyden, E.S.; et al. Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929–943.e928.
- Tycko, R. Molecular Structure of Aggregated Amyloid-β: Insights from Solid-State Nuclear Magnetic Resonance. Cold Spring Harb. Perspect. Med. 2016, 6, a024083.
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513.
- Condello, C.; Stöehr, J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 2018, 109, 191–200.
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312.
- Pignataro, A.; Middei, S. Trans-Synaptic Spread of Amyloid-β in Alzheimer’s Disease: Paths to β-Amyloidosis. Neural Plast. 2017, 2017, 5281829.
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334.
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216.
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35.
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345.
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470.
- Funato, H.; Yoshimura, M.; Yamazaki, T.; Saido, T.C.; Ito, Y.; Yokofujita, J.; Okeda, R.; Ihara, Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 1998, 152, 983–992.
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967.
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896.
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434.
- Li, Z.; Wang, N.; Yue, T.; Liu, L. Matrine reverses the drug resistance of K562/ADM cells to ADM and VCR via promoting autophagy. Transl. Cancer Res. 2020, 9, 786–794.
- Choi, Y.; Kim, H.S.; Shin, K.Y.; Kim, E.M.; Kim, M.; Kim, H.S.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.P.; et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 2393–2404.
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101.
- Kolb, B.; Whishaw, I.Q. Fundamentals of Human Neuropsychology; Macmillan: New York, NY, USA, 2009.
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027.
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Bajetto, A.; Bonavia, R.; Barbero, S.; Schettini, G. Characterization of chemokines and their receptors in the central nervous system: Physiopathological implications. J. Neurochem. 2002, 82, 1311–1329.
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34.
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2017, 27, 795–808.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217.
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56.
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171.
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
11 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No