Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anthony A Grace | -- | 2215 | 2023-08-09 18:20:37 | | | |
| 2 | Sirius Huang | Meta information modification | 2215 | 2023-08-10 04:29:34 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Grace, A.A.; Uliana, D.L. Mechanism of Action of D2 Antagonist Antipsychotic Medications. Encyclopedia. Available online: https://encyclopedia.pub/entry/47852 (accessed on 26 July 2026).
Grace AA, Uliana DL. Mechanism of Action of D2 Antagonist Antipsychotic Medications. Encyclopedia. Available at: https://encyclopedia.pub/entry/47852. Accessed July 26, 2026.
Grace, Anthony A., Daniela L. Uliana. "Mechanism of Action of D2 Antagonist Antipsychotic Medications" Encyclopedia, https://encyclopedia.pub/entry/47852 (accessed July 26, 2026).
Grace, A.A., & Uliana, D.L. (2023, August 09). Mechanism of Action of D2 Antagonist Antipsychotic Medications. In Encyclopedia. https://encyclopedia.pub/entry/47852
Grace, Anthony A. and Daniela L. Uliana. "Mechanism of Action of D2 Antagonist Antipsychotic Medications." Encyclopedia. Web. 09 August, 2023.
Copy Citation
Therapeutic intervention for schizophrenia relies on blockade of dopamine D2 receptors in the associative striatum; however, there is little evidence for baseline overdrive of the dopamine system. Instead, the dopamine system is in a hyper-responsive state due to excessive drive by the hippocampus. This causes more dopamine neurons to be in a spontaneously active, hyper-responsive state. Antipsychotic drugs alleviate this by causing depolarization block, or excessive depolarization-induced dopamine neuron inactivation.
dopamine
antipsychotic
schizophrenia
1. Introduction
Schizophrenia is a devastating developmental disorder that arises from an interaction of genetic susceptibility and environmental factors [1][2][3][4], with an incidence of approximately 1–1.5% worldwide [5][6][7]. The illness has a devastating impact on individuals and their families/caregivers [8], striking during late adolescence/early adulthood [5][9]. As a result, there has been substantial effort dedicated to providing a better understanding of the pathophysiology of this disorder as a means to discover more effective treatments. The first treatment developed was based on dopamine antagonism. This was a serendipitous discovery that was made on the basis of an opportune observation that a modified antihistamine, chlorpromazine, was an effective stabilizer for extended surgical interventions [10][11]. In coining the term “neuroleptic” to describe the neural stabilizing action of the drug, Laborit [12] attracted the attention of clinicians working in a mental asylum as a potential pharmacological intervention for a number of at that time untreatable mental disorders. They found that the drug was highly effective for those experiencing schizophrenia [13][14][15][16][17]. It was another 20 years before the drug was proposed to exert its therapeutic action via blockade of dopamine (DA) receptors [18][19], and another 12 years before the DA type 2 receptor (D2) was identified as the binding site of antipsychotic drugs [20]. This, along with evidence of DA releasing drugs exacerbating schizophrenia psychosis [21][22][23], has given rise to the DA hypothesis of schizophrenia [24][25], which has dominated drug development in the treatment of schizophrenia for decades.
2. Mechanism of Action of D2 Antagonist Antipsychotic Medications
A breakthrough came from animal studies of the actions of repeated antipsychotic drug administration on the DA system. In recordings from identified DA neurons in the brains of rats, it was found that 3+ weeks of treatment with first-generation antipsychotic drugs led to an inactivation of DA neuron firing—a phenomenon known as depolarization block [26][27][28]. It was found that the first-generation drugs caused depolarization block in both the nigrostriatal and mesolimbic DA system [26][27][29][30]. The nigrostriatal dopaminergic pathway projecting from substantia nigra compacta to the dorsal striatum is known to regulate motor control, and the mesolimbic pathway regulates projecting from the ventral tegmental area to the ventral striatum and limbic system, which are linked to reward, motivation, and emotion [31]. Interestingly, the second-generation drugs, which did not have extrapyramidal side effects but still exhibited therapeutic efficacy, only caused depolarization block in the mesolimbic psychosis-related DA system but not in the extrapyramidal nigrostriatal DA system [32][33][34]. Finally, it was shown that a compound with limited antipsychotic actions but prominent extrapyramidal actions, metoclopramide [35], only produced depolarization block in the nigrostriatal system but not the mesolimbic system [33]. Therefore, it was established that repeated antipsychotic drug-induced depolarization block of the mesolimbic system was associated with antipsychotic efficacy, and depolarization block of the nigrostriatal system with extrapyramidal side effects [29][33][34]. While this was a powerful correlation, there were some caveats: (1) antipsychotic drugs do not need to be administered for weeks to obtain therapeutic efficacy in patients, with onset being observed within 24 h of drug treatment [36]; (2) there was still no evidence for a hyperactive DA system; and (3) DA neuron depolarization block is not the normal state of the DA system.
One potential issue with these preclinical experiments is that they were performed on normal rats. It is well known that the brain has extensive homeostatic mechanisms that can compensate for the continued presence of a drug, and it was likely that the delayed action in the normal animals may have been due to these compensatory mechanisms [37]. What about animal models of schizophrenia, in which the system is already disrupted? The researchers have proposed that in a disrupted system, in which homeostatic processes are either disrupted or ineffective, a therapeutic agent would have significantly different actions as compared to a normal system [37]. For this reason, it is essential to test these agents in an animal model that can approximate at least some of the circuit disruptions that clinical studies have found to also be altered in the schizophrenia brain [37][38].
One issue in this approach is identifying an appropriate animal model. While it is clear that one cannot precisely replicate a complex uniquely human disorder like schizophrenia in a rodent, one can use clinical studies to guide development of an appropriate model and evaluate its functional validity. Evidence shows that schizophrenia likely has a strong genetic component, with the heritability of schizophrenia a function of number of shared genes in families [1][2]. In addition, there is clear evidence that environmental factors can also engender susceptibility to schizophrenia [1][3][4]. Furthermore, studies by Weinberger and others have shown that disruption of hippocampal function early in life will result in a rodent model that can recapitulate a number of features of schizophrenia [39][40][41][42][43][44]. In particular, imaging studies in schizophrenia patients have revealed hyperactivity in the limbic hippocampus [45], which is associated with an increase fluorodopa uptake in the associative striatum [46][47]. The associative striatum is the area of the striatum that receives inputs from associative areas of the neocortex and which correspond anatomically to the medial caudate-putamen segments of the striatum [48][49][50]. Similarly, in the MAM rats, researchers observed hyperactivity in the ventral limbic hippocampus and an increase in ventral tegmental area DA neuron activity [51]. This increase in DA neuron activity would thus be consistent with the increased fluorodopa uptake, since fluorodopa uptake is a metric of the number of active terminals [46][52], and researchers observed an increase in the number of DA neurons driving these terminals [52][53]. The hyperactivity of the hippocampus appears to be driven by a loss of parvalbumin-containing gamma-aminobutyric acidergic (GABAergic) interneurons, which is observed in postmortem schizophrenia brains [54][55] as well as in the MAM model of schizophrenia [56][57]. Furthermore, researchers showed that activation of the ventral hippocampus would, through a circuit involving the nucleus accumbens and the ventral pallidum, lead to an increase in the number of active DA neurons in the ventral tegmentum [58] (Figure 1). Thus, researchers propose that the overactive DA system involved in schizophrenia psychosis is due to loss of parvalbumin inhibition of the ventral hippocampus and consequent overdrive of the DA system. Importantly, while the overdriven DA system is likely the source of the psychotic positive symptoms of schizophrenia [59][60], the limbic hippocampus also exhibits projections to areas involved in affective regulation (e.g., the amygdala) and cognition (i.e., the prefrontal cortex) [61][62][63] (Figure 2). Thus, while targeting the DA system may help to relieve psychosis, it will not impact the negative and cognitive symptoms of this disorder.
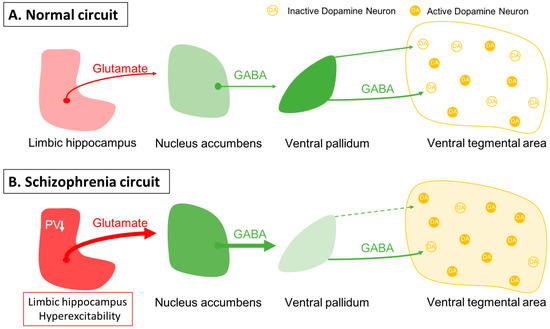
Figure 1. Dopamine neuron activity is driven by a pacemaker conductance that maintains their firing, which is offset by inhibition from the ventral pallidum. (A) In the baseline state, approximately half of the dopamine neurons are spontaneously firing, with the other half held in a hyperpolarized state due to GABAergic inhibition from the ventral pallidum. Activity in the limbic hippocampus provides an excitatory drive to the nucleus accumbens, which in turn can inhibit the ventral pallidum to modulate the inhibitory drive on ventral tegmental area dopamine neurons [58]. (B) In schizophrenia, a loss of parvalbumin GABAergic neurons in the limbic hippocampus causes this region to be tonically hyperactive; this leads to increased accumbens inhibition of the ventral pallidum. This releases the dopamine neurons from inhibition, causing the entire population of dopamine neurons to be in the active, responsive state.
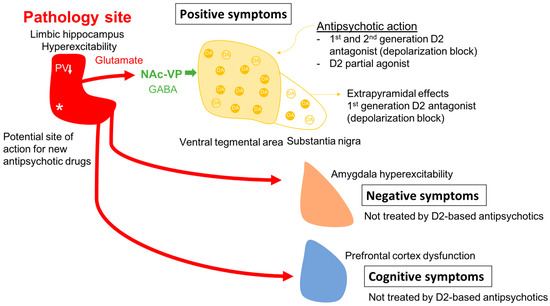
Figure 2. In schizophrenia, a loss of parvalbumin GABAergic neurons in the limbic hippocampus, via a circuit involving the nucleus accumbens and ventral pallidum leads to a disinhibition of VTA dopamine neurons and dopamine overdrive in the associative striatum; this appears to underlie the positive psychotic features of the disorder. By causing depolarization block of the ventral tegmental dopamine neurons, first- and second-generation antipsychotic drugs reverse dopamine neuron hyperactivity to relieve psychosis; in addition, first-generation drugs also cause depolarization block in the substantia nigra dopamine neurons to lead to extrapyramidal side effects. However, when the hippocampus is hyperactive and dysrhythmic, it can also lead to pathological activity changes in its other targets. Thus, it will impact the amygdala-cingulate cortex to lead to negative symptoms, and the prefrontal cortex to induce cognitive deficits; all characteristics of schizophrenia that are not effectively treated by dopamine antagonist first- and second-generation antipsychotic drugs. Novel mechanism antipsychotic drugs that act directly at the site of pathology in the hippocampus could be effective at reversing the negative and cognitive deficits as well as the positive symptoms of schizophrenia. * Site of pathology in schizophrenia; ↓ PV reduction in the hippocampus.
Given that the DA neurons are not hyperactive individually, but instead there are more of the neurons active, this would cause a stimulus that increases DA neuron firing and would have a substantially greater effect. Specifically, if the DA system is hyper-responsive to stimuli, this would cause an inappropriately high DA response to what may be benign stimuli, a condition known as aberrant salience [64][65][66]. This would lead to inappropriate attribution of threat or salience to benign stimuli (i.e., delusions) (Figure 3).
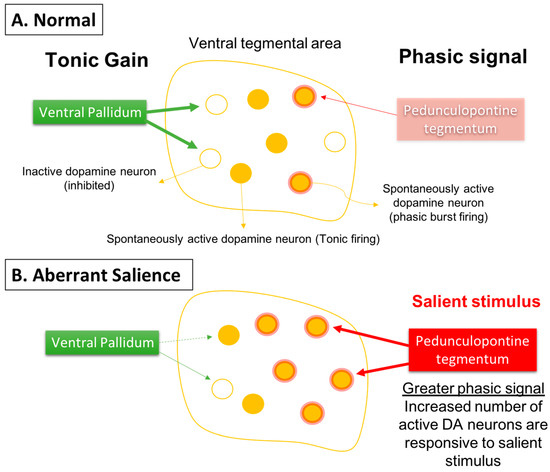
Figure 3. Phasic dopamine neuron burst firing is believed to be the behaviorally salient output of the dopamine system. (A) Burst firing is driven by a glutamate input from the pedunculopontine tegmentum acting on dopamine neuron n-methyl-D-aspartate (NMDA) receptors to drive burst firing. However, for NMDA to drive burst firing, the neuron must be in a depolarized, spontaneously active state; otherwise in hyperpolarized, inactive neurons there is a magnesium blockade of the NMDA channel. Therefore, only spontaneously active dopamine neurons can be driven by the pedunculopontine tegmentum into burst firing. The ventral pallidum, by controlling the number of dopamine neurons active, can determine the level of amplification, or the gain, of the phasic response. This is thought to be adjusted depending on the demands of the environment; in highly salient or dangerous conditions, the hippocampus increases the number of dopamine neurons in the responsive, active state, thereby enabling a salient stimulus to activate the pedunculopontine tegmental-driven burst firing across a large number of dopamine neurons, facilitating an immediate response to the threat. (B) In the case of schizophrenia, an overactive hippocampus removes tonic inhibitory drive of dopamine neurons, causing a massive increase in the number of responsive neurons independent of environmental contingencies. Under these conditions, both salient and nonsalient stimuli will cause a maximal phasic response. Therefore, with an overdriven dopamine system, every stimulus will be perceived as a threat, causing the patient to be overwhelmed and unable to filter salient from nonsalient stimuli. This leads to a state of aberrant salience, or the inappropriate attribution of salience to a normally benign object.
How would D2 antagonists help to alleviate this condition? In the normal animal, administration of antipsychotic drugs, by blocking postsynaptic D2 receptors, would cause a feedback activation of the DA system that, over weeks, results in DA neuron depolarization block. However, in the schizophrenia patient, the DA system is already in a hyperactive state [28]. In the MAM rats, researchers found that administration of a D2 antagonist, unlike in a normal animal, would add to the present hyper-responsive DA system, with the result that depolarization block develops very soon after drug administration [67]. This is consistent with the clinical literature, with the antipsychotic properties produced rapidly after antipsychotic medication initiation. Moreover, the more psychotic the patient, the more rapid the onset of therapeutic action [36][68]. Again, this is consistent with the MAM rat, in that the more overdriven the DA system is initially, the more rapidly will addition of a D2 antagonist produce depolarization block. By producing depolarization block and inactivation of DA neuron firing, this would alleviate the pathological increase in the number of DA neurons active and hyper-responsiveness to stimuli.
This provides a circuit-based assessment of the mechanism of antipsychotic drug treatment alleviation of psychosis [69]. However, it also exposes several caveats: (1) the “normal” state of the DA system is not depolarization block and this may be why antipsychotic drugs produce negative affective states, which may contribute to low patient compliance [70]; and (2) this will impact the DA system not by treating schizophrenia at the site of pathology (i.e., the limbic hippocampus), but instead 5 synapses downstream from the defect [66]. Thus, while the D2 blocking antipsychotic drug may alleviate psychosis, it would be ineffective in treating negative and cognitive deficits of this disorder.
How can some of these issues be circumvented? One more recent development is the use of partial DA agonist drugs, such as aripiprazole or brexpiprazole [71]. These drugs will occupy D2 receptors but, rather than producing a complete blockade, will produce a partial activation of the receptor [72]. Thus, these drugs will act by preventing overstimulation of D2 receptors while providing a baseline level of stimulation. For this reason, these drugs are typically administered at doses that occupy D2 receptors approximately 95% without producing excessive blockade-induced negative affect. The researchers found that, unlike first- and second-generation antipsychotic drugs [28], the partial agonist aripiprazole appears to directly inhibit DA neuron activity without inducing depolarization block [73]. Therefore, while an improvement over receptor blockade-induced depolarization block, the drugs will nonetheless fail to alleviate the negative and cognitive deficits associated with schizophrenia, and will still impact the normal function of the DA system.
References
- Miller, P.; Lawrie, S.M.; Hodges, A.; Clafferty, R.; Cosway, R.; Johnstone, E.C. Genetic liability, illicit drug use, life stress and psychotic symptoms: Preliminary findings from the Edinburgh study of people at high risk for schizophrenia. Soc. Psychiatry Psychiatr. Epidemiol. 2001, 36, 338–342.
- Henriksen, M.G.; Nordgaard, J.; Jansson, L.B. Genetics of Schizophrenia: Overview of Methods, Findings and Limitations. Front. Hum. Neurosci. 2017, 11, 322.
- Popovic, D.; Schmitt, A.; Kaurani, L.; Senner, F.; Papiol, S.; Malchow, B.; Fischer, A.; Schulze, T.G.; Koutsouleris, N.; Falkai, P. Childhood Trauma in Schizophrenia: Current Findings and Research Perspectives. Front. Neurosci. 2019, 13, 274.
- Berthelot, N.; Garon-Bissonnette, J.; Jomphe, V.; Doucet-Beaupré, H.; Bureau, A.; Maziade, M. Childhood trauma may increase risk of psychosis and mood disorder in genetically high-risk children and adolescents by enhancing the accumulation of risk indicators. Schizophr. Bull. Open 2022, 3, sgac017.
- McCutcheon, R.A.; Reis Marques, T.; Howes, O.D. Schizophrenia—An Overview. JAMA Psychiatry 2020, 77, 201.
- Messias, E.L.; Chen, C.-Y.; Eaton, W.W. Epidemiology of schizophrenia: Review of findings and myths. Psychiatr. Clin. N. Am. 2007, 30, 323–338.
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global Epidemiology and Burden of Schizophrenia: Findings from the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203.
- Kadakia, A.; Catillon, M.; Fan, Q.; Williams, G.R.; Marden, J.R.; Anderson, A.; Kirson, N.; Dembek, C. The Economic Burden of Schizophrenia in the United States. J. Clin. Psychiatry 2022, 83, 22m14458.
- Gomes, F.V.; Rincón-Cortés, M.; Grace, A.A. Adolescence as a period of vulnerability and intervention in schizophrenia: Insights from the MAM model. Neurosci. Biobehav. Rev. 2016, 70, 260–270.
- Ban, T.A. Fifty years chlorpromazine: A historical perspective. Neuropsychiatr. Dis. Treat. 2007, 3, 495–500.
- Shen, W.W. A history of antipsychotic drug development. Compr. Psychiatry 1999, 40, 407–414.
- Laborit, H.; Huguenard, P. Artificial hibernation by pharmacodynamic and physical means, in surgery. J. Chir. 1951, 67, 631–641.
- Casey, J.F.; Bennett, I.F.; Lindley, C.J.; Hollister, L.E.; Gordon, M.H.; Springer, N.N. Drug therapy in schizophrenia. A controlled study of the relative effectiveness of chlorpromazine, promazine, phenobarbital, and placebo. AMA Arch. Gen. Psychiatry 1960, 2, 210–220.
- Elkes, J.; Elkes, C. Effect of chlorpromazine on the behavior of chronically overactive psychotic patients. Br. Med. J. 1954, 2, 560–565.
- Lehmann, H.E.; Hanrahan, G.E. Chlorpromazine; new inhibiting agent for psychomotor excitement and manic states. AMA Arch. Neurol. Psychiatry 1954, 71, 227–237.
- Delay, J.; Deniker, P.; Harl, J.M. Therapeutic method derived from hiberno-therapy in excitation and agitation states. Ann. Med. Psychol. 1952, 110, 267–273.
- Hamon; Paraire; Velluz. Effect of R. P. 4560 on maniacal agitation. Ann. Med. Psychol. 1952, 110, 331–335.
- Creese, I.; Burt, D.R.; Snyder, S.H. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 1976, 192, 481–483.
- Seeman, P.; Lee, T.; Chau-Wong, M.; Wong, K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature 1976, 261, 717–719.
- Seeman, P. Dopamine receptor sequences. Therapeutic levels of neuroleptics occupy D2 receptors, clozapine occupies D4. Neuropsychopharmacology 1992, 7, 261–284.
- Abi-Dargham, A.; Van De Giessen, E.; Slifstein, M.; Kegeles, L.S.; Laruelle, M. Baseline and Amphetamine-Stimulated Dopamine Activity Are Related in Drug-Naïve Schizophrenic Subjects. Biol. Psychiatry 2009, 65, 1091–1093.
- Laruelle, M.; Abi-Dargham, A.; Van Dyck, C.H.; Gil, R.; D’Souza, C.D.; Erdos, J.; McCance, E.; Rosenblatt, W.; Fingado, C.; Zoghbi, S.S.; et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. USA 1996, 93, 9235–9240.
- Laruelle, M.; Abi-Dargham, A.; Gil, R.; Kegeles, L.; Innis, R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol. Psychiatry 1999, 46, 56–72.
- Gründer, G.; Cumming, P. The Dopamine Hypothesis of Schizophrenia. In The Neurobiology of Schizophrenia; Elsevier: Amsterdam, The Netherlands, 2016; pp. 109–124. ISBN 978-0-12-801829-3.
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.-G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front. Psychiatry 2014, 5, 47.
- Bunney, B.S.; Grace, A.A. Acute and chronic haloperidol treatment: Comparison of effects on nigral dopaminergic cell activity. Life Sci. 1978, 23, 1715–1727.
- Grace, A.A.; Bunney, B.S. Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: Analysis using in vivo intracellular recording. J. Pharmacol. Exp. Ther. 1986, 238, 1092–1100.
- Grace, A.A.; Bunney, B.S.; Moore, H.; Todd, C.L. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997, 20, 31–37.
- Grace, A.A. The depolarization block hypothesis of neuroleptic action: Implications for the etiology and treatment of schizophrenia. J. Neural. Transm. Suppl. 1992, 36, 91–131.
- Lane, R.F.; Blaha, C.D. Chronic haloperidol decreases dopamine release in striatum and nucleus accumbens in vivo: Depolarization block as a possible mechanism of action. Brain Res. Bull 1987, 18, 135–138.
- Luo, S.X.; Huang, E.J. Dopaminergic Neurons and Brain Reward Pathways: From Neurogenesis to Circuit Assembly. Am. J. Pathol. 2016, 186, 478–488.
- Chiodo, L.A.; Bunney, B.S. Typical and atypical neuroleptics: Differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. J. Neurosci. 1983, 3, 1607–1619.
- White, F.J.; Wang, R.Y. Differential effects of classical and atypical antipsychotic drugs on A9 and A10 dopamine neurons. Science 1983, 221, 1054–1057.
- Goldstein, J.M.; Litwin, L.C.; Sutton, E.B.; Malick, J.B. Seroquel: Electrophysiological profile of a potential atypical antipsychotic. Psychopharmacology 1993, 112, 293–298.
- Harrington, R.A.; Hamilton, C.W.; Brogden, R.N.; Linkewich, J.A.; Romankiewicz, J.A.; Heel, R.C. Metoclopramide. An updated review of its pharmacological properties and clinical use. Drugs 1983, 25, 451–494.
- Kapur, S.; Arenovich, T.; Agid, O.; Zipursky, R.; Lindborg, S.; Jones, B. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am. J. Psychiatry 2005, 162, 939–946.
- Uliana, D.L.; Gomes, F.V.; Grace, A.A. Update on current animal models for schizophrenia: Are they still useful? Curr. Opin. Psychiatry 2023, 36, 172–178.
- Uliana, D.L.; Zhu, X.; Gomes, F.V.; Grace, A.A. Using animal models for the studies of schizophrenia and depression: The value of translational models for treatment and prevention. Front. Behav. Neurosci. 2022, 16, 935320.
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669.
- Lipska, B.K.; Jaskiw, G.E.; Weinberger, D.R. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: A potential animal model of schizophrenia. Neuropsychopharmacology 1993, 9, 67–75.
- Chambers, R.A.; Moore, J.; McEvoy, J.P.; Levin, E.D. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology 1996, 15, 587–594.
- Becker, A.; Grecksch, G.; Bernstein, H.G.; Höllt, V.; Bogerts, B. Social behaviour in rats lesioned with ibotenic acid in the hippocampus: Quantitative and qualitative analysis. Psychopharmacology 1999, 144, 333–338.
- Grecksch, G.; Bernstein, H.G.; Becker, A.; Höllt, V.; Bogerts, B. Disruption of latent inhibition in rats with postnatal hippocampal lesions. Neuropsychopharmacology 1999, 20, 525–532.
- Tseng, K.Y.; Chambers, R.A.; Lipska, B.K. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav. Brain Res. 2009, 204, 295–305.
- Heckers, S.; Konradi, C. Hippocampal pathology in schizophrenia. Curr. Top Behav. Neurosci. 2010, 4, 529–553.
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The Nature of Dopamine Dysfunction in Schizophrenia and What This Means for Treatment: Meta-analysis of Imaging Studies. Arch. Gen. Psychiatry 2012, 69, 776–786.
- McGowan, S.; Lawrence, A.D.; Sales, T.; Quested, D.; Grasby, P. Presynaptic Dopaminergic Dysfunction in Schizophrenia: A Positron Emission Tomographic Fluorodopa Study. Arch. Gen. Psychiatry 2004, 61, 134.
- Joel, D.; Weiner, I. The organization of the basal ganglia-thalamocortical circuits: Open interconnected rather than closed segregated. Neuroscience 1994, 63, 363–379.
- Parent, A.; Hazrati, L.-N. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res. Rev. 1995, 20, 91–127.
- Joel, D.; Weiner, I. The connections of the dopaminergic system with the striatum in rats and primates: An analysis with respect to the functional and compartmental organization of the striatum. Neuroscience 2000, 96, 451–474.
- Lodge, D.J.; Grace, A.A. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 2007, 27, 11424–11430.
- Modinos, G.; Allen, P.; Grace, A.A.; McGuire, P. Translating the MAM model of psychosis to humans. Trends Neurosci. 2015, 38, 129–138.
- Moore, H.; Jentsch, J.D.; Ghajarnia, M.; Geyer, M.A.; Grace, A.A. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: Implications for the neuropathology of schizophrenia. Biol. Psychiatry 2006, 60, 253–264.
- Zhang, Z.J.; Reynolds, G.P. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr. Res. 2002, 55, 1–10.
- Konradi, C.; Yang, C.K.; Zimmerman, E.I.; Lohmann, K.M.; Gresch, P.; Pantazopoulos, H.; Berretta, S.; Heckers, S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr. Res. 2011, 131, 165–173.
- Lodge, D.J.; Behrens, M.M.; Grace, A.A. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. 2009, 29, 2344–2354.
- Gill, K.M.; Grace, A.A. Corresponding decrease in neuronal markers signals progressive parvalbumin neuron loss in MAM schizophrenia model. Int. J. Neuropsychopharmacol. 2014, 17, 1609–1619.
- Floresco, S.B.; Todd, C.L.; Grace, A.A. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J. Neurosci. 2001, 21, 4915–4922.
- Wong, D.F.; Wagner, H.N.; Tune, L.E.; Dannals, R.F.; Pearlson, G.D.; Links, J.M.; Tamminga, C.A.; Broussolle, E.P.; Ravert, H.T.; Wilson, A.A.; et al. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science 1986, 234, 1558–1563.
- Tost, H.; Alam, T.; Meyer-Lindenberg, A. Dopamine and psychosis: Theory, pathomechanisms and intermediate phenotypes. Neurosci. Biobehav. Rev. 2010, 34, 689–700.
- Ghoshal, A.; Conn, P.J. The hippocampo-prefrontal pathway: A possible therapeutic target for negative and cognitive symptoms of schizophrenia. Future Neurol. 2015, 10, 115–128.
- O’Donnell, P.; Grace, A.A. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: Hippocampal gating of prefrontal cortical input. J. Neurosci. 1995, 15, 3622–3639.
- O’Donnell, P.; Lewis, B.L.; Weinberger, D.R.; Lipska, B.K. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb. Cortex 2002, 12, 975–982.
- Kapur, S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 2003, 160, 13–23.
- Sonnenschein, S.F.; Gomes, F.V.; Grace, A.A. Dysregulation of Midbrain Dopamine System and the Pathophysiology of Schizophrenia. Front. Psychiatry 2020, 11, 613.
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532.
- Gill, K.M.; Cook, J.M.; Poe, M.M.; Grace, A.A. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophr. Bull. 2014, 40, 341–350.
- Haddad, P.M.; Correll, C.U. The acute efficacy of antipsychotics in schizophrenia: A review of recent meta-analyses. Ther Adv Psychopharmacol. 2018, 8, 303–318.
- Sonnenschein, S.F.; Grace, A.A. Insights on current and novel antipsychotic mechanisms from the MAM model of schizophrenia. Neuropharmacology 2020, 163, 107632.
- Haddad, P.M.; Brain, C.; Scott, J. Nonadherence with antipsychotic medication in schizophrenia: Challenges and management strategies. Patient Relat. Outcome Meas. 2014, 5, 43–62.
- Mohr, P.; Masopust, J.; Kopeček, M. Dopamine Receptor Partial Agonists: Do They Differ in Their Clinical Efficacy? Front. Psychiatry 2022, 12, 781946.
- Burris, K.D.; Molski, T.F.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, F.D.; Molinoff, P.B. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002, 302, 381–389.
- Sonnenschein, S.F.; Gill, K.M.; Grace, A.A. State-dependent effects of the D2 partial agonist aripiprazole on dopamine neuron activity in the MAM neurodevelopmental model of schizophrenia. Neuropsychopharmacology 2019, 44, 572–580.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
10 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No