Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Monika Konaklieva | -- | 3121 | 2023-08-03 16:42:25 | | | |
| 2 | Sirius Huang | Meta information modification | 3121 | 2023-08-04 08:54:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Konaklieva, M.I.; Plotkin, B.J. Microbe-Specific Targeting by β-Lactams and Ureas. Encyclopedia. Available online: https://encyclopedia.pub/entry/47628 (accessed on 25 June 2026).
Konaklieva MI, Plotkin BJ. Microbe-Specific Targeting by β-Lactams and Ureas. Encyclopedia. Available at: https://encyclopedia.pub/entry/47628. Accessed June 25, 2026.
Konaklieva, Monika I., Balbina J. Plotkin. "Microbe-Specific Targeting by β-Lactams and Ureas" Encyclopedia, https://encyclopedia.pub/entry/47628 (accessed June 25, 2026).
Konaklieva, M.I., & Plotkin, B.J. (2023, August 03). Microbe-Specific Targeting by β-Lactams and Ureas. In Encyclopedia. https://encyclopedia.pub/entry/47628
Konaklieva, Monika I. and Balbina J. Plotkin. "Microbe-Specific Targeting by β-Lactams and Ureas." Encyclopedia. Web. 03 August, 2023.
Copy Citation
β-Lactams have been viewed as universal acylating agents of serine and cysteine enzymes of both prokaryotic and eukaryotic systems. Their use has been propelled by the COVID-19 pandemic, thus broadening their application as inhibitors of viral enzymes. The urea-based drugs have been extensively studied as inhibitors of the aforementioned enzymes.
β-lactams
ureas
antimicrobials
antivirals
enzyme inhibitors
1. Introduction
The structural features of amides have been utilized by nature for millennia. One of the most successful examples of naturally occurring amide-based enzyme products targeting proteins is the group of β-lactams. For close to a century, the β-lactams have served as a rich source of marketed drugs and have opened new areas of target-based drug discovery. They are representatives of the mechanism-based covalent modification of a target enzyme as a key step in achieving potent inhibition of that target. They are also an example of how nature utilizes small molecules to modify key catalytic residues at an enzyme active site. Newly developed covalent modulators of non-catalytic amino acid residues have also been successful as anticancer drugs.
More recently, there has been a turn in the direction of drug development towards the utilization of ureas as targeted covalent enzyme inhibitors. Originally, ureas were marginalized by organic/medicinal chemistry since they lacked the natural ubiquity of amides, which were the historical focus of enzyme inhibition research. However, after a slow start as antimicrobials, initiated by Paul Ehrlich, urea-based inhibitors have gained traction in the past twenty years as HIV and other viral inhibitors, as well as anticancer drugs.
The focus herein will be on the general electrophile–nucleophile interaction utilized by β-lactams and ureas and their bioisosters in the context of developing anti-infectives in the current age of rapid microbial resistance growth.
-
General Chemistry and Structure
Most current β -lactams that target microbes modify their targets, mainly proteases, and are viewed as universal acylating agents of prokaryotic and eukaryotic serine and cysteine enzymes through covalent modification [1][2][3]. These targets and mechanisms of action are shared by a broad range of β-lactam structures, with penicillin being the first-identified antimicrobial of this drug class. The value of the β-lactam ring, a small heterocycle which features a reactive electrophilic center, has been demonstrated for a multitude of molecular targets (Figure 1).
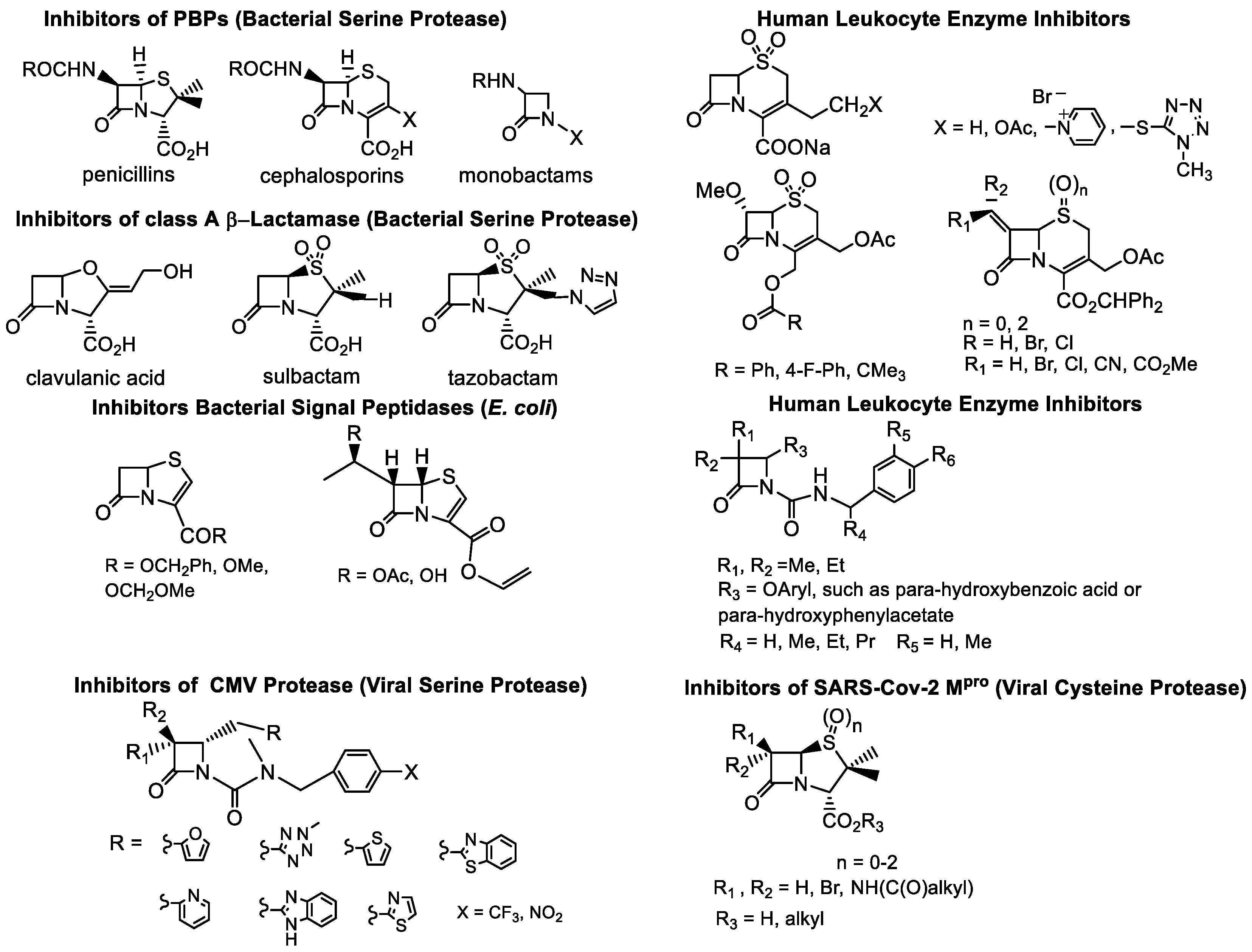
Figure 1. Illustrative examples of the “classical” β-lactams: acylating agents of proteases.
There are also other examples, such as clinically relevant Ezetimibe (Figure 2), intended as an inhibitor (acylating agent) of one of the enzymes associated with the mammalian lipid metabolism (Acyl-CoA:cholesterol acyltransferase, ACAT); however, its molecular target has not yet been completely identified. Currently, Ezetimibe’s mechanism of action is accepted to be an inhibition of a transport protein target called Niemann-Pick C1- Like 1 (NPC1L1) [4][5]. Although non-acylating β-lactams, e.g., alkylthio-β-lactams (Figure 2), also contain the β-lactam functionality, their anti-microbial activity it is not based on enzyme-acylating, but on S-alkylation of the bacterial coenzyme A (CoA) [6][7]. The focus herein will be on the acylating agents, which are the largest β-lactam class developed to target infectious agents (Figure 1).
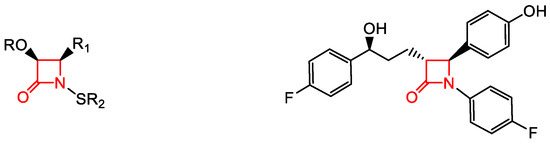
Figure 2. Illustrative examples of β-lactams: interfering with bacterial fatty acid synthesis, N-alkylthio β-lactams and adsorption of mammalian cholesterol, Ezetimide, respectively.
-
Lactams—Cyclic Amides
The stability and acylating function of a simple β-lactam ring is comparable to that of simple acyclic amide. Briefly, the carbonyl group of either the acyclic amide, or its cyclic counterpart, e.g., the β-lactam, engages in hydrogen bonding in the so-called “oxyanion” hole of a given enzyme. This binding allows for stabilization of the negative charge on the oxygen atom of the carbonyl, which secures the general acid–base catalysis, namely an attack of a nucleophile, most often a primary alcohol, such as the one from serine on the amide carbonyl carbon, as the electrophile. The nucleophilic attack on the latter leads to the formation of a tetrahedral intermediate, a species whose collapse results in acylation of the enzyme. This covalent binding of an amide/β-lactam with an enzyme has essential requirements, which have been extensively described in the literature [1][8][9].
Overall, β-lactams exhibit good pharmacokinetics/pharmacodynamics. Thus, they continue to be on the forefront of recently approved antimicrobial drugs, with the newest being the “pan”-antimicrobial cephalosporin Cefiderocol (Figure 3). This cephalosporin is the only one out of the twelve antimicrobials approved in the past five years, with activity against critical class pathogens as defined by the World Health Organization (WHO) [10]. Cefiderocol has a catechol moiety in its side chain which acts as an iron chelator. This allows Cefiderol to be actively transported across the cell membrane via iron transporter channels [11]. This transport process avoids current resistance mechanisms associated with passive diffusion. Furthermore, Cefiderocol is minimally affected by multidrug efflux pumps and is stable against hydrolysis by β-lactamases, including metallo-β-lactamases [11][12]. This ability to bypass several major mechanisms of bacterial resistance makes Cefiderocol suitable for the treatment of Gram-negative bacteria [11][12].
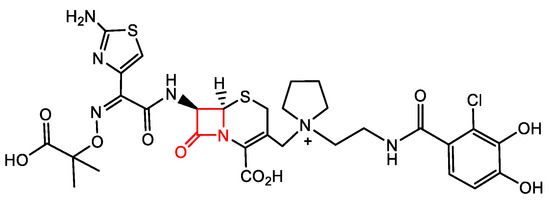
Figure 3. The latest FDA-approved pan-antimicrobial, Cefiderocol.
There are a number of other amide-based clinically approved acyclic amides and β-lactam drugs, including lidocaine, paracetamol, β-lactam antibiotics, and β-lactamase inhibitors, e.g., penicillin, clavulanic acid, and monocyclic β-lactams, as well as compounds with various other activities, e.g., ezetimibe, atorvastatin, chloramphenicol, moclobemide, captopril, acetazolamide, ponatinib, methotrexate, and trimethobenzamide. Although the best-known mode of action for β-lactam antibiotics is through inhibition of cell wall synthesis, their activity has also been demonstrated to interfere with bacterial quorum sensing [13]. In addition to their activity against prokaryotes, they have been shown to regulate innate immune responses through their effect on myeloid cells [14].
-
Bioisosters of Amides—Ureas
A short-fall of amide-based structures is that they have limited stability in vivo. Therefore, use of these compounds requires replacing the initial amide-based lead compound with a peptide mimic, i.e., a bioisoster. There are different heterocyclic molecules that are broadly used in drug design as amide bioisosters. Their properties are elegantly described in recent perspectives and reviews [15][16][17]. The focus herein is on the urea-based scaffold commonly used in drug development for the treatment of bacterial and viral infections.
The earliest example of a urea compound developed as an anti-infective having anti-parasitic (anti-trypanosome) activity was developed by the Nobel laureate Paul Ehrlich (Figure 4). Recently, some compounds that were developed for mammalian (human) diseases (e.g., cancer) have been re-purposed as anti-microbials, including anti-virals, via exploitation of the mammalian metabolic pathways that are hijacked by the viruses.
Figure 4. Trypan Red analog—the molecule at the foundation of modern chemotherapy (Paul Ehrlich sought for the first time to correlate the chemical structure of a synthetic drug with its biological effects).
The ability of urea to form stable hydrogen bonds with a drug target is the core quality that allows for specificity in drug action. Modification of the physicochemical properties of the molecule by increasing its stability also leads to the development of new drugs [15][16]. It is the use of various types of substituents on the urea nitrogen atoms that significantly alters the conformational preferences of the urea derivatives as they appear to have a wide variety of drug targets [16]. Ureas typically are more tempered electrophiles (as compared to β-lactams) covalently inhibiting individual serine hydrolases with excellent potency and selectivity [18][19][20][21]. Ureas’ utility as antimicrobials are being further expanded through the testing of currently FDA-approved urea-based drugs which have potential for drug repurposing. These drugs are anticancer agents (e.g., the kinase inhibitors Sorafenib and Regorafenib, Figure 5), stearoyl-CoA desaturase 1, as well as inhibitors of the insulin-like growth factor I receptor.
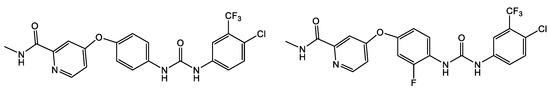
Figure 5. Urea-based FDA-approved kinase inhibitors Sorafenib and Regorafenib (anti-cancer agents), respectively.
2. Microbe-Specific Targeting by β-Lactams and Ureas
2.1. Gram-Positive Staphylococcus aureus and Enterococcus faecalis
Despite the increasing resistance to antibiotics, β-lactams, particularly the anti-staphylococcal penicillins (ASPs), e.g., nafcillin, oxacillin, and flucloxacillin and the first-generation cephalosporins, e.g., cefazolin, remain the primary treatments for methicillin-sensitive Staphylococcus aureus (MSSA) infections, including bacteremia and infective endocarditis, although with restrictions [22][23]. An overview of the structure–function relationship for β-lactams and ureas with activity against Staphylococcus aureus and Enterococcus faecalis is shown in Table 1. Similarly, Enterococcus faecalis clinical isolates are resistant against most of the clinically relevant β-lactams with only the third- and fourth-generation cephalosporins and the carbapenems remaining that are efficacious. Monocyclic β-lactams substituted on the lactam nitrogen with the sulphonyl benzene pharmacophore were prepared recently in search of novel antimicrobial and antiviral compounds [24]. Compound 1 (Table 1) demonstrated one of the best antimicrobial activities of all compounds tested against both Gram-positive and Gram-negative organisms, with an MIC of 1 μg/mL for S. aureus. The N-phenylsulfonyl ureas (e.g., 1, Table 1) have moderate antiviral activity, but are not promising enough for further optimization of the scaffold as antivirals [24].
Beyond involvement with penicillin binding proteins (PBP) as the mode of action, kinase inhibitors, such as Sorafenib and Regorafenib (Figure 5, Table 6) FDA-approved anticancer agents), have demonstrated antibacterial and antiviral activity. The Sorafenib analog PK150 (Table 1) has demonstrated antimicrobial activity against MSSA strains and a 10-fold enhanced anti-MRSA activity. In addition, PK150 is as effective in the elimination of pre-existing biofilms as urea 2 (Table 1) with better activity than either Sorafenib or Regorafenib (3 µM for both Sorafenib and Regorafenib) [25]. Additionally, PK150 is an inhibitor of demethylmenaquinone methyltranferase (MenG) biosynthesis, which is involved in S. aureus menaquinone metabolism. Furthermore, it is not affected by environmentally acquired resistance as it causes over-activation of SpsB, an S. aureus signal peptidase I enzyme [25]. PK150 also shows oral bioavailability and antibacterial activity against several pathogenic strains at submicromolar concentrations [25].
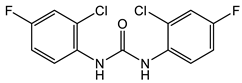
Triclocarban belongs to the same chemotype as Sorafenib and Regorafenib, but was FDA approved specifically as an antimicrobial. Triclocarban’s mechanism of action, which is similar to Sorafenib, has MenG as its molecular target [26]. However, due to its numerous side effects, and its lack of timely degradation in the environment, it was banned by the FDA in 2016 for use in hygiene consumer products [27][28][29]. The development of triclocarban analogs with reduced toxicity led to the development of a series of diarylureas, some of which can dissipate pre-existing S. aureus biofilms. The most promising diarylurea in this library is the thiofluoro-substituted urea 2 (Table 1) that has increased selectivity against MRSA (MIC, 0.05 μg/mL) [30].
Other substitutions include PQ 401 (Table 1), an inhibitor of insulin-like growth factor I receptor (IGF-1R) signaling, which is important in both breast cancer and osteosarcoma [31]. PQ 401, which has demonstrated bactericidal activity against S. aureus strains, including MRSA, functions by disruption of the lipid bilayer [32]. Meanwhile, incorporating an aminoguanidine group into the diarylurea scaffold, such as in compounds 3 and 4 (Table 1), results in compounds that interfere with cell wall biosynthesis and produce re-sensitization of vancomycin-resistant S. aureus (VRSA) to vancomycin [33]. In high concentrations, these guanidinolated ureas also have S. aureus anti-biofilm activity [34]. Alternatively, substituting one of the aryl groups in the diarylureas with a pyrazoyl group led to the preparation of pyrazoyl-ureas 5 (Table 1) [35]. These compounds, when evaluated for antimicrobial activity against S. aureus, E. coli, and B. subtilis, demonstrated a moderate bacteriostatic/bacteriocidal activity of 250 μg/mL for all organisms tested. Furthermore, pyrazoyl-substituted ureas of type 6 (Table 1) demonstrated good antimicrobial activity against both Gram-positive and Gram-negative bacteria with S. aureus MICs in the 0.8 mg/mL range for both the trifluoromethyl- and chloro-substituted aromatic ring [36]. The pyrazoyl-substituted diarylureas have additional activities, such as antifungal and anti-malarial activity [37]. Diarylureas 7 and 8 also demonstrate activity against Enterococcus faecalis (MIC = 31.3 µg/mL, Table 1) and Klebsiella pneumoniae (MIC = 31.3 µg/mL, Table 2). Interestingly, both 7 and 8 are the only ones from the ureas described that contain a hydroxy group and a Michael acceptor. Whether this chemical difference affects their activity against these organisms remains to be answered [38]. In all cases, a disubstituted urea is necessary for activity and active compounds require, in addition to the pyrazole scaffold, an N-aryl substituent preferably 3,4 or 3,5-disubstituted with halogen atoms and/or CF3 groups. Unfortunately, these ureas demonstrate low bioavailability in vivo, a result that could be attributed to low solubility [36].
Table 1. Representative examples of Gram-positive bacteria with recognized/reported β-lactams and ureas as antimicrobials: Staphylococcus aureus and Enterococcus faecalis. The structures of the lactams and ureas associated with a given microorganism with their corresponding references (bolded, in parentheses) are shown.
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| S. aureus | 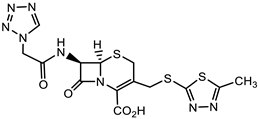 Cephazolin (first-generation cephalosporin) [22] 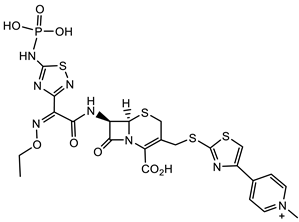 Ceftaroline (first-generation cephalosporin) [23] 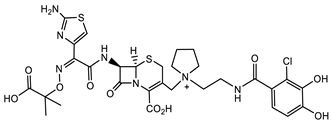 Cefiderocol (approved for medical use in the United States in November 2019, and in the European Union in April 2020) [10][11][12] 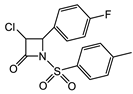 1 [24] MIC = 1 mg/mL |
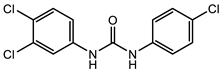 Triclocarban (TCC) [26][27][28][29] 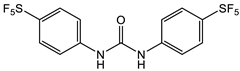 2 [25][30] S. aureus (ATCC, MIC50 = 0.5 µg/mL) as well as MRSA (MIC50 = 0.05 µg/mL) 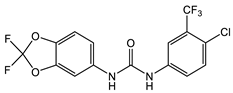 PK150 [25] MIC 0.3 µM for MSSA, MIC 0.03 µM for MRSA. 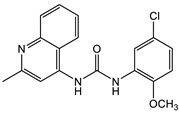 PQ 401 [31][32] |
| MRSA/ Biofilm |
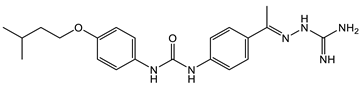 3 [33][34] 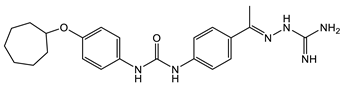 4 [33][34] MIC values for 3 and 4 between 2.0 and 4.0 µg/mL as compared with vancomycin (from 0.5 to 1 µg/mL). Molecular target: cell wall synthesis |
|
| S. aureus | 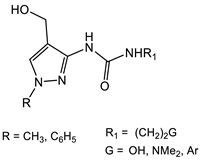 5 [35] MIC = 250 mg/mL Compound 5 was evaluated as an antibacterial and antifungal agent against Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 25922), Bacillus subtilis (ATCC 8236F800), and Candida albicans (ATCC 885-653), showing a moderate antimicrobial activity. 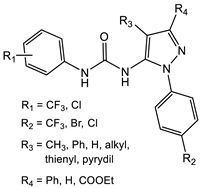 6 [36] IC50 ranging from 0.054 to 4.24 μM |
|
| E. faecalis |  Cefepime—fourth-generation cephalosporin [39]  Meropenem [40] |
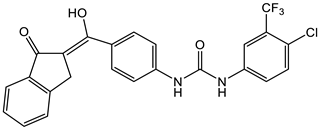
 7 [38] 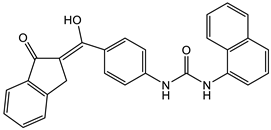 8 [38] |
2.2. Gram-Negative Multi-Drug Resistant Bacteria: Proteus mirabilis, Klebsiella pneumoniae, and Acinetobacter baumannii
Resistance to β-lactam drugs is even more pronounced with respect to Gram-negative pathogenic bacteria, such as Proteus mirabilis, Klebsiella pneumoniae, and Acinetobacter baumannii. While some strains of P. mirabilis are sensitive to ampicillin, in general, broad-spectrum penicillins (e.g., ticarcillin and piperacillin), first-, second-, and third-generation cephalosporins (e.g., imipenem and aztreonam), carbapenems, and piperacillin/tazobactam are required for use in the treatment of extended-spectrum β-lactamase-producing P. mirabilis bacteremia [39]. With respect to the treatment of K. pneumoniae infections, the use of β-lactams is limited to third- and fourth-generation cephalosporins and carbapenems. The only clinically used monobactam, aztreonam, has good activity against many Gram-negative bacteria; however, its activity against some of the most problematic multidrug-resistant (MDR) strains of P. aeruginosa and A. baumannii is limited [40]. Interestingly, coupling of the aztreonam side chain free acid with a bis-catechol siderophore mimetic (lactam 9, Table 2) significantly improves the activity against the MDR strains of Gram-negative bacteria that are of most significant concern [41].
With regard to the activity of diarylureas, there are several representatives of this class of compounds with activity against Gram-negative bacteria. The chloro, fluoro-, and trifluorpmethyl-substituted diarylureas 10 and 11 (Table 2) demonstrated activity against P. mirabilis that is comparable to that of ciprofloxacin [42]. In compound 12 (Table 2), one of the aryl groups of the prototypic diarylurea was substituted by an adamantane group, a feature that increases the lipophilicity of the compound [43]. Compound 12 (Table 2) demonstrated very good selectivity against A. baumannii (94% inhibition at 32 μg/mL) as compared with the rest of the bacteria tested, such as S. aureus, K. pneumonia, E. coli, P. aeruginosa, and C. albicans. Using several other analogs containing the adamantane group, it was determined that these compounds bind to the active site of enoyl-(acyl-carrier-protein) reductase (ENR). Of the library of adamantine-based analogs screened, compound 12 (Table 2) remains the best compound against A. baumannii, with analogs of diarylureas 7 and 8 (Table 1 and Table 2) exhibiting good activity against E. faecalis (MIC = 31.3 µg/mL, Table 1) and K. pneumoniae (MIC = 31.3 µg/mL, Table 2).
Table 2. Representative examples of Gram-negative bacteria with recognized/reported β-lactams and ureas as antimicrobials: Proteus mirabilis, Klebsiella pneumoniae, Acinetobacter baumannii. The structures of the lactams and ureas associated with a given microorganism with their corresponding references (bolded, in parentheses) are shown.
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| P. mirabilis | 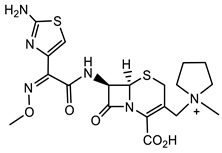 Cefepime [39] fourth-generation cephalosporin 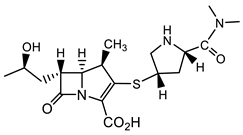 Meropenem [39] |
 10 [42] 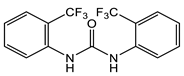 11 [42] |
| K. pneumoniae | 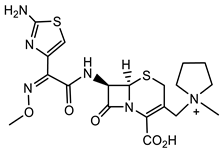 Cefepime [40] fourth-generation cephalosporin 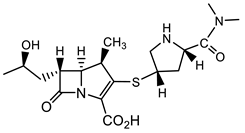 Meropenem [40] 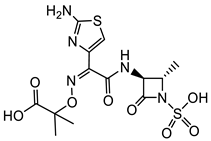 Aztreonam [41] |
 7 [38] 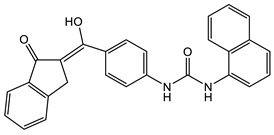 8 [38] |
| A. baumannii | 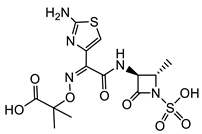 Aztreonam [41] 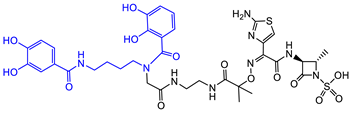 9 [41] Aztreonam with siderophore mimetic |
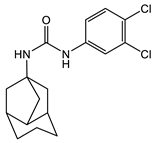 12 [43] |
2.3. Mycobacterium tuberculosis (Mtb)
Until over a decade ago, Mycobacterium tuberculosis (Mtb) was considered refractory to β-lactam antibiotics. However, more recent studies show that carbapenems have anti-Mtb activity and are now used in the treatment of disease caused by MDR and extremely drug-resistant (XDR) Mtb strains [44][45]. Following this recognition of efficacy, further drug screening (8900 β-lactams) by a consortium of pharmaceutical companies, the NIH, and academic institutions identified about 1600 compounds that exhibited activity against Mtb, including many minus the necessity for the presence of a β-lactamase inhibitor [46]. Representative structures of cephalosporins from this work (lactam 13, which is active against replicating (R), and 14, which is active against both R and non-replicating (NR) Mtb) are shown in Table 3 [46]. Identification of β-lactams that are cidal for both R and NR Mtb was an additional reason to propel this massive screen undertaking [46][47]. Among the cephalosporins developed, six have a pyrithione-leaving group and are dual-acting compounds with activity against both NR and R Mtb. The release of the pyrithione upon cleavage of the β-lactam ring by β-lactamases, and/or engagement of the β-lactam ring with its target, allows for these two parts of the molecule to contribute to antitubercular activity.
A new family of anti-Mtb agents consists of C4-phenylthio β-lactams with the 15 scaffold (Table 3) [48]. This family shares similarities to the monocyclic structures 20 (Table 4) with the majority of structures having a benzylthio substituent at C4 and a urea moiety at N1 [49]. Lactams such as 15 having a phenylthio group at C4, and a urea moiety at N1, have shown activity against R and NR Mtb, as well as in macrophages [48]. Lactams with the 20 scaffold (Table 4) have antiviral activity as inhibitors of human cytomegalovirus (HCMV) protease with acceptable anti-HCMV activity [49]. Improvements upon the physicochemical properties of the monocyclic β-lactams 15 (Table 3) in laboratories are ongoing.
Table 3. Representative examples with recognized/reported β-lactams and ureas as antimicrobials: Mycobacterium tuberculosis (Mtb). The structures of the lactams and ureas associated with Mtb and the corresponding references (bolded, in parentheses) are shown.
| Microorg. | Structure—β-Lactam/Reference | Structure—Urea/Reference |
|---|---|---|
| Mtb | 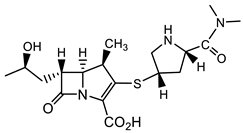 Meropenem [44][45] 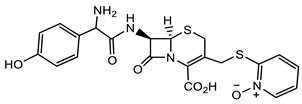 13 [46][47] 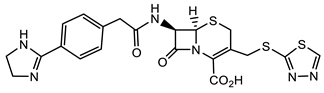 14 [46][47] (TBBL-0000316) 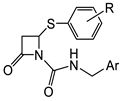 15 [48] R = o-F, MIC 1.5 µg/mL (M. tuberculosis pathogenic strain H37Rv) |
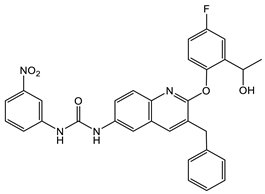 16 [50] MIC 6.25 µg/mL (M. tuberculosis pathogenic strain H37Rv) 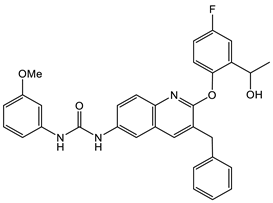 17 [50] MIC 3.125 µg/mL (M. tuberculosis pathogenic strain H37Rv) 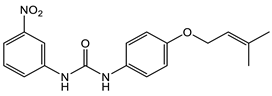 18 [31] MIC 6.0 µg/mL (M. tuberculosis pathogenic strain H37Rv) 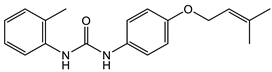 19 [31] MIC 5.2 µg/mL (M. tuberculosis pathogenic strain H37Rv) |
Nitro-substituted diarylurea 16 and methoxy-substituted diarylurea 17 (Table 3) inhibited Mtb H37Rv with an MIC of 6.25 μg/mL and 3.12 μg/mL, respectively [50]. These MIC values are higher than those of isoniazid (0.05–0.2 μg/mL), the most often used drug as a control for evaluation of anti-Mtb activity, but are encouraging for further compound development. Nitro-substituted diarylurea 18 and methyl-substituted diaryl urea 19 (Table 3) have similar MIC values of 6.0 and 5.2 µg/mL, respectively, to compounds 16 and 17 (Table 3) against Mtb H37Rv. Interestingly, these diaryl-substituted ureas proved to be selective inhibitors of mycolic acid biosynthesis [51].
In summary, the predominant development of antimicrobials continues to be focused on the bacterial molecular targets exploring dual action, e.g., anti-Mtb inhibitors of both the PBPs and β-lactamases. Cefiderocol is the only antibiotic, out of the 12 approved since 2017, able to target all the pathogens considered critical by the WHO. The rest of the antibiotics are under development in Phase 1 to 3 clinical trials. As of this year (2023), from a total of 27 compounds, only four have new mechanisms of action, only six are considered innovative enough to meet the WHO criteria for the treatment of bacteria-resistant organisms, and only two of these six target the most difficult organisms to treat, such as A. baumannii and P. aeruginosa. Most of the drugs under development are the next generation of existing classes.
References
- Fisher, J.F.; Mobashery, S. Chapter 3: The β-Lactam (Azetidin-2-one) as a Privileged Ring in Medicinal Chemistry. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; Bräse, S., Ed.; Royal Chemical Society of Chemictry, RSC Drug Discovery, 2015; Available online: https://books.rsc.org/books/edited-volume/529/Privileged-Scaffolds-in-Medicinal-Chemistry-Design (accessed on 21 March 2023).
- Konaklieva, M.I.; Plotkin, B.J. The relationship between inhibitors of eukaryotic and prokaryotic serine proteases. Mini Rev. Med. Chem. 2004, 4, 721–739.
- Kluge, A.F.; Petter, R.S. Acylating drugs: Redesigning natural covalent inhibitors. Curr. Opin. Chem. Biol. 2010, 14, 421–427.
- Burnett, D.A. β-Lactam cholesterol absorption inhibitors. Curr. Med. Chem. 2004, 11, 1873–1887.
- Garcia-Calvo, M.; Lisnock, J.-M.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137.
- Revell, K.D.; Heldreth, B.; Long, T.E.; Jang, S.; Turos, E. N-thiolated β-lactams: Studies on the mode of action and identification of a primary cellular target in Staphylococcus aureus. Bioorg. Med. Chem. 2007, 15, 2453–2467.
- Konaklieva, M.I. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics 2014, 3, 128–142.
- Fisher, J.F.; Mobashery, S. β-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev. 2021, 121, 3412–3463.
- Fisher, J.F.; Mobashery, S. β-Lactams from the Ocean. Mar. Drugs 2023, 21, 86.
- WHO: More New Antibiotics Needed. Available online: https://www.idse.net/Bacterial-Infections/Article/03-23/WHO-More-New-Antibiotics-Needed/69730 (accessed on 21 March 2023).
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A.; et al. Cefiderocol: A siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-negative bacilli. Drugs 2019, 79, 271–289.
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543.
- Kumar, L.; Brenner, N.; Brice, J.; Klein-Seetharaman, J.; Sarkar, S.K. Cephalosporins Interfere with Quorum Sensing and Improve the Ability of Caenorhabditis elegans to Survive Pseudomonas aeruginosa Infection. Front. Microbiol. 2021, 12, 598498.
- Mondeme, M.; Carnoy, C.; Sirard, J.-C.; Faveeuw, C. Treatment of Bacterial Infections with β-Lactams: Cooperation with Innate Immunity. Infect. Immun. 2023, 2, 1–11.
- Kumari, S.; Carmona, A.V.; Towari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358.
- Ghosh, A.K.; Brindisi, M. Urea Derivatives in Modern Drug Discovery and Medicinal Chemistry. J. Med. Chem. 2020, 63, 2751–2788.
- Ronchetti, R.; Moroni, G.; Carotti, A.; Gioiello, A.; Camaioni, E. Recent advances in urea- and thiourea-containing compounds: Focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 2021, 12, 1046.
- Adibekian, A.; Martin, B.R.; Wang, C.; Hsu, K.-L.; A Bachovchin, D.; Niessen, S.; Hoover, H.; Cravatt, B.F. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol. 2011, 7, 469–478.
- Chang, J.W.; Nomura, D.K.; Cravatt, B.F. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol. 2011, 18, 476–484.
- Hsu, K.L.; Tsuboi, K.; Adibekian, A.; Pugh, H.; Masuda, K.; Cravatt, B.F. DAGLbeta inhibition perturbs a lipid network involved in macrophage infammatory responses. Nat. Chem. Biol. 2012, 8, 999–1007.
- Remsberg, J.R.; Suciu, R.M.; Zambetti, N.A.; Hanigan, T.W.; Firestone, A.J.; Inguva, A.; Long, A.; Ngo, N.; Lum, K.M.; Henry, C.L.; et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat. Chem. Biol. 2021, 17, 856–864.
- Li, J.; Echevarria, K.L.; Traugott, K.A. β-Lactam therapy for methicillin-susceptible Staphylococcus aureus bacteremia: A comparative review of cefazolin versus antistaphylococcal penicillins. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 346–360.
- Dingle, T.C.; Gamage, D.; Gomez-Villegas, S.; Hanson, B.M.; Reyes, J.; Abbott, A.; Burnham, C.-A.D.; Bard, J.D.; Fritz, S.; Miller, W.R.; et al. Prevalence and Characterization of the Cefazolin Inoculum Effect in North American Methicillin-Susceptible Staphylococcus aureus Isolates. J. Clin. Microbiol. 2022, 60.
- Mandal, M.K.; Ghosh, S.; Bhat, H.R.; Naesens, L.; Singh, U.P. Synthesis and biological evaluation of substituted phenyl azetidine-2-one sulphonyl derivatives as potential antimicrobial and antiviral agents. Bioorg. Chem. 2020, 104, 104320.
- Le, P.; Kunold, E.; Macsics, R.; Rox, K.; Jennings, M.C.; Ugur, I.; Reinecke, M.; Chaves-Moreno, D.; Hackl, M.W.; Fetzer, C.; et al. Repurposing human kinase inhibitors to create an antibiotic active against drug-resistant Staphylococcus aureus, persisters and biofilms. Nat. Chem. 2020, 12, 145–158.
- Macsics, R.; Hackl, M.W.; Fetzer, C.; Mostert, D.; Bender, J.; Layer, F.; Sieber, S.A. Comparative Target Analysis of Chlorinated Biphenyl Antimicrobials Highlights MenG as Molecular Target of Triclocarban. Appl. Environ. Microbiol. 2020, 86.
- FDA Issues Final Rule on Safety and Effectiveness of Antibacterial Soaps. Available online: https://www.fda.gov/news-events/press-announcements/fda-issues-final-rule-safety-and-effectiveness-antibacterial-soaps (accessed on 12 February 2023).
- Wang, Y.; Teng, Y.; Wang, D.; Han, K.; Wang, H.; Kang, L. The fate of triclocarban in activated sludge and its influence on biological wastewater treatment system. J. Environ. Manag. 2020, 276, 111237.
- Catalano, A.; Iacopetta, D.; Pellegrino, M.; Aquaro, S.; Franchini, C.; Sinicropi, M.S. Diarylureas: Repositioning from Antitumor to Antimicrobials or Multi-Target Agents against New Pandemics. Antibiotics 2021, 10, 92.
- Pujol, E.; Blanco-Cabra, N.; Julián, E.; Leiva, R.; Torrents, E.; Vázquez, S. Pentafluorosulfanyl-containing triclocarban analogs with potent antimicrobial activity. Molecules 2018, 23, 2853.
- Catalano, A.; Iacopetta, D.; Sinicropi, M.S.; Franchini, C. Diarylureas as antitumor agents. Appl. Sci. 2021, 11, 374.
- Kim, W.; Zou, G.; Pan, W.; Fricke, N.; Faizi, H.A.; Kim, S.M.; Khader, R.; Li, S.; Lee, K.; Escorba, L.; et al. The neutrally charged diarylurea compound PQ401 kills antibiotic-resistant and antibiotic-tolerant Staphylococcus aureus. Mbio 2020, 11, 603.
- Eissa, I.H.; Mohammad, H.; Qassem, O.A.; Younis, W.; Abdelghany, T.M.; Elshafeey, A.; Moustafa, M.M.A.R.; Seleem, M.N.; Mayhoub, A.S. Diphenylurea derivatives for combating methicillin- and vancomycin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2017, 130, 73–85.
- Mohammad, H.; Younis, W.; Ezzat, H.G.; Peters, C.E.; AbdelKhalek, A.; Cooper, B.; Pogliano, K.; Pogliano, J.; Mayhoub, A.S.; Seleem, M.N. Bacteriological profiling of diphenylureas as a novel class of antibiotics against methicillin-resistant Staphylococcus aureus. PLoS ONE 2017, 12, e0182821.
- Bratenko, M.K.; Barus, M.M.; Rotar, D.V.; Vovk, M.V. Polyfunctional Pyrazoles. 9*. Synthesis of 1-Alkyl(Aryl)-3-ureas. Chem. Heterocyc. Compd. 2014, 50, 1252–1258.
- Kane, J.L.; Hirth, B.H.; Liang, B.; Gourlie, B.B.; Nahill, S.; Barsomian, G. Ureas of 5-aminopyrazole and 2-aminothiazole inhibit growth of gram-positive bacteria. Bioorg. Med. Chem. Lett. 2003, 13, 4463–4466.
- Brullo, C.; Rapetti, F.; Bruno, O. Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules 2020, 25, 3457.
- Gezegen, H.; Hepokur, C.; Tutar, U.; Ceylan, M. Synthesis and Biological Evaluation of Novel 1-(4-(Hydroxy(1-oxo-1,3-dihydro-2H-inden-2-ylidene)methyl)phenyl)-3-phenylurea Derivatives. Chem. Biodivers. 2017, 14, e1700223.
- Jamil, R.T.; Foris, L.A.; Snowden, J. Proteus Mirabilis Infections. Available online: https://www.ncbi.nlm.nih.gov/books/NBK442017/ (accessed on 30 March 2023).
- Ashurst, J.V.; Dawson, A. Klebsiella Pneumoniae. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519004/ (accessed on 30 March 2023).
- Liu, R.; Miller, P.A.; Miller, M.J. Conjugation of Aztreonam, a Synthetic Monocyclic β-Lactam Antibiotic, to a Siderophore Mimetic Significantly Expands Activity Against Gram-Negative Bacteria. ACS Infect. Dis. 2021, 7, 2979–2986.
- Sarveswari, S.; Vijayakumar, V. A Facile Synthesis of diaylureas and their antimicrobial evaluation. Chiang Mai J. Sci. 2018, 45, 997–1007.
- Patil, M.; Noonikara-Poyil, A.; Joshi, S.D.; Patil, S.A.; Patil, S.A.; Bugarin, A. New Urea Derivatives as Potential Antimicrobial Agents: Synthesis, Biological Evaluation, and Molecular Docking Studies. Antibiotics 2019, 8, 178.
- Diacon, A.H.; van der Merwe, L.; Barnard, M.; von Groote-Bidlingmaier, F.; Lange, C.; García-Basteiro, A.L.; Sevene, E.; Ballell, L.; Barros-Aguirre, D. β-Lactams against Tuberculosis--New Trick for an Old Dog? N. Engl. J. Med. 2016, 375, 393–394.
- van Rijn, S.P.; Zuur, M.A.; Anthony, R.; Wilffert, B.; van Altena, R.; Akkerman, O.W.; de Lange, W.C.M.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.C. Evaluation of Carbapenems for Treatment of Multi- and Extensively Drug-Resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e01489-18.
- Gold, B.; Zhang, J.; Quezada, L.L.; Roberts, J.; Ling, Y.; Wood, M.; Shinwari, W.; Goullieux, L.; Roubert, C.; Fraisse, L.; et al. Identification of β-Lactams Active against Mycobacterium tuberculosis by a Consortium of Pharmaceutical Companies and Academic Institutions. ACS Infect. Dis. 2022, 8, 557–573.
- Lopez Quezada, L.; Li, K.; McDonald, S.L.; Nguyen, Q.; Perkowski, A.J.; Pharr, C.W.; Gold, B.; Roberts, J.; McAulay, K.; Saito, K.; et al. Dual-Pharmacophore Pyrithione-Containing Cephalosporins Kill Both Replicating and Nonreplicating Mycobacterium tuberculosis. ACS Infect. Dis. 2019, 5, 1433–1445.
- Kuskovsky, R.; Lloyd, D.; Arora, K.; Plotkin, B.; Green, J.; Boshoff, H.; Barry, C.; Deschamps, J.; Konaklieva, M.I. C4-Phenylthio β-lactams: Effect of the Chirality of the β-Lactam Ring on Antimicrobial Activity. Bioorg. Med. Chem. 2019, 27, 115050.
- Déziel, R.; Malenfant, E. Inhibition of human cytomegalovirus protease No with monocyclic β-lactams. Bioorg. Med. Chem. Lett. 1998, 8, 1437–1442.
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V. Chattapadhayaya, Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 4681–4692.
- Velappan, A.B.; Raja, M.R.C.; Datta, D.; Tsai, Y.T.; Halloum, I.; Wan, B.; Kremer, L.; Gramajo, H.; Franzblau, S.G.; Mahapatra, S.K.; et al. Attenuation of Mycobacterium species through direct and macrophage mediated pathway by unsymmetrical diaryl urea. Eur. J. Med. Chem. 2017, 125, 825–841.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
671
Revisions:
2 times
(View History)
Update Date:
04 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No