 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emma Sierecki | -- | 2751 | 2023-08-02 03:37:35 | | | |
| 2 | Dean Liu | Meta information modification | 2751 | 2023-08-03 02:25:23 | | |
Video Upload Options
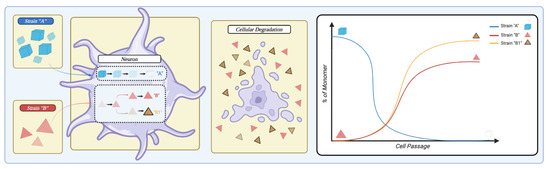
Like many neurodegenerative diseases, Parkinson’s disease (PD) is characterized by the formation of proteinaceous aggregates in brain cells. In PD, those proteinaceous aggregates are formed by the α-synuclein (αSyn) and are considered the trademark of this neurodegenerative disease. In addition to PD, αSyn pathological aggregation is also detected in atypical Parkinsonism, including Dementia with Lewy Bodies (DLB), Multiple System Atrophy (MSA), as well as neurodegeneration with brain iron accumulation, some cases of traumatic brain injuries, and variants of Alzheimer’s disease. Collectively, these (and other) disorders are referred to as synucleinopathies, highlighting the relation between disease type and protein misfolding/aggregation. Despite these pathological relationships, however, synucleinopathies cover a wide range of pathologies, present with a multiplicity of symptoms, and arise from dysfunctions in different neuroanatomical regions and cell populations. Strikingly, αSyn deposition occurs in different types of cells, with oligodendrocytes being mainly affected in MSA, while aggregates are found in neurons in PD. If multiple factors contribute to the development of a pathology, especially in the cases of slow-developing neurodegenerative disorders, the common presence of αSyn aggregation, as both a marker and potential driver of disease, is puzzling.
1. The Strain-to-Cell Connection
2. Cell-to-Strain Imprinting
References
- Courte, J.; Bousset, L.; Von Boxberg, Y.; Villard, C.; Melki, R.; Peyrin, J.M. The Expression Level of Alpha-Synuclein in Different Neuronal Populations Is the Primary Determinant of Its Prion-like Seeding. Sci. Rep. 2020, 10, 1–13.
- Tanudjojo, B.; Shaikh, S.S.; Fenyi, A.; Bousset, L.; Agarwal, D.; Marsh, J.; Zois, C.; Heman-Ackah, S.; Fischer, R.; Sims, D.; et al. Phenotypic Manifestation of α-Synuclein Strains Derived from Parkinson’s Disease and Multiple System Atrophy in Human Dopaminergic Neurons. Nat. Commun. 2021, 12, 3817.
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169.
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841.
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Agid, Y.; Dürr, A.; Brice, A.; Pollak, P. Causal Relation between α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1169–1171.
- Eriksen, J.L.; Przedborski, S.; Petrucelli, L. Gene Dosage and Pathogenesis of Parkinson’s Disease. Trends Mol. Med. 2005, 11, 91–96.
- Choi, Y.R.; Park, S.J.; Park, S.M. Molecular Events Underlying the Cell-to-Cell Transmission of α-Synuclein. FEBS J. 2021, 288, 6593–6602.
- Lee, H.-J.; Suk, J.-E.; Bae, E.-J.; Lee, J.-H.; Paik, S.R.; Lee, S.-J. Assembly-Dependent Endocytosis and Clearance of Extracellular Alpha-Synuclein. Int. J. Biochem. 2008, 40, 1835–1849.
- Janda, E.; Boi, L.; Carta, A.R. Microglial Phagocytosis and Its Regulation: A Therapeutic Target in Parkinson’s Disease? Front. Mol. Neurosci. 2018, 11, 44.
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C.; et al. Mechanistic Basis for Receptor-Mediated Pathological α-Synuclein Fibril Cell-to-Cell Transmission in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118.
- Uemura, N.; Uemura, M.T.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med. 2020, 26, 936–952.
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological α-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science 2016, 353, aah3374.
- Gu, H.; Yang, X.; Mao, X.; Xu, E.; Qi, C.; Wang, H.; Brahmachari, S.; York, B.; Sriparna, M.; Li, A.; et al. Lymphocyte Activation Gene 3 (Lag3) Contributes to α-Synucleinopathy in α-Synuclein Transgenic Mice. Front. Cell Neurosci. 2021, 15, 656426.
- Emmenegger, M.; De Cecco, E.; Hruska-Plochan, M.; Eninger, T.; Schneider, M.M.; Barth, M.; Tantardini, E.; de Rossi, P.; Bacioglu, M.; Langston, R.G.; et al. LAG3 Is Not Expressed in Human and Murine Neurons and Does Not Modulate A-synucleinopathies. EMBO Mol. Med. 2021, 13, e14745.
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147.
- Hudák, A.; Kusz, E.; Domonkos, I.; Jósvay, K.; Kodamullil, A.T.; Szilák, L.; Hofmann-Apitius, M.; Letoha, T. Contribution of Syndecans to Cellular Uptake and Fibrillation of α-Synuclein and Tau. Sci. Rep. 2019, 9, 16543.
- Leonova, E.I.; Galzitskaya, O.V. The Role of Syndecan-2 in Amyloid Plaque Formation. Mol. Biology. Maik Nauka 2015, 49, 89–98.
- Letoha, T.; Hudák, A.; Kusz, E.; Pettkó-Szandtner, A.; Domonkos, I.; Jósvay, K.; Hofmann-Apitius, M.; Szilák, L. Contribution of Syndecans to Cellular Internalization and Fibrillation of Amyloid-β(1–42). Sci. Rep. 2019, 9, 1–17.
- Stopschinski, B.E.; Holmes, B.B.; Miller, G.M.; Manon, V.A.; Vaquer-Alicea, J.; Prueitt, W.L.; Hsieh-Wilson, L.C.; Diamond, M.I. Specific Glycosaminoglycan Chain Length and Sulfation Patterns Are Required for Cell Uptake of Tau versus α-Synuclein and β-Amyloid Aggregates. J. Biol. Chem. 2018, 293, 10826–10840.
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C.; et al. α-Synuclein Assemblies Sequester Neuronal A3-Na+/K+-ATPase and Impair Na+ Gradient. EMBO J. 2015, 34, 2408–2423.
- Dzamko, N.; Gysbers, A.; Perera, G.; Bahar, A.; Shankar, A.; Gao, J.; Fu, Y.H.; Halliday, G.M. Toll-like Receptor 2 Is Increased in Neurons in Parkinson’s Disease Brain and May Contribute to Alpha-Synuclein Pathology. Acta Neuropathol. 2017, 133, 303–319.
- Feng, Y.; Zheng, C.; Zhang, Y.; Xing, C.; Cai, W.; Li, R.; Chen, J.; Duan, Y. Triptolide Inhibits Preformed Fibril-Induced Microglial Activation by Targeting the MicroRNA155-5p/SHIP1 Pathway. Oxid. Med. Cell Longev. 2019, 2019, 1–13.
- Xia, Y.; Zhang, G.; Kou, L.; Yin, S.; Han, C.; Hu, J.; Wan, F.; Sun, Y.; Wu, J.; Li, Y.; et al. Reactive Microglia Enhance the Transmission of Exosomal α-Synuclein via Toll-like Receptor 2. Brain 2021, 144, 2024–2037.
- Shrivastava, A.N.; Bousset, L.; Renner, M.; Redeker, V.; Savistchenko, J.; Triller, A.; Melki, R. Differential Membrane Binding and Seeding of Distinct α-Synuclein Fibrillar Polymorphs. Biophys. J. 2020, 118, 1301–1320.
- Neupane, S.; De Cecco, E.; Aguzzi, A. The Hidden Cell-to-Cell Trail of α-Synuclein Aggregates. J. Mol. Biol. 2023, 435, 167930.
- Raghav, A.; Singh, M.; Jeong, G.B.; Giri, R.; Agarwal, S.; Kala, S.; Gautam, K.A. Extracellular Vesicles in Neurodegenerative Diseases: A Systematic Review. Front. Mol. Neurosci. 2022, 15, 1061076.
- Meloni, M.; Agliardi, C.; Guerini, F.R.; Zanzottera, M.; Bolognesi, E.; Picciolini, S.; Marano, M.; Magliozzi, A.; Di Fonzo, A.; Arighi, A.; et al. Oligomeric α-Synuclein and Tau Aggregates in NDEVs Differentiate Parkinson’s Disease from Atypical Parkinsonisms. Neurobiol. Dis. 2023, 176, 105947.
- Hernandez, S.M.; Tikhonova, E.B.; Karamyshev, A.L. Protein-Protein Interactions in Alpha-Synuclein Biogenesis: New Potential Targets in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 72.
- Stephens, A.D.; Zacharopoulou, M.; Moons, R.; Fusco, G.; Seetaloo, N.; Chiki, A.; Woodhams, P.J.; Mela, I.; Lashuel, H.A.; Phillips, J.J.; et al. Extent of N-Terminus Exposure of Monomeric Alpha-Synuclein Determines Its Aggregation Propensity. Nat. Commun. 2020, 11, 2820.
- Landureau, M.; Redeker, V.; Bellande, T.; Eyquem, S.; Melki, R. The Differential Solvent Exposure of N-Terminal Residues Provides “Fingerprints” of Alpha-Synuclein Fibrillar Polymorphs. J. Biol. Chem. 2021, 296, 100737.
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575.
- Suzuki, G.; Imura, S.; Hosokawa, M.; Katsumata, R.; Nonaka, T.; Hisanaga, S.-I.; Saeki, Y.; Hasegawa, M. α-Synuclein Strains That Cause Distinct Pathologies Differentially Inhibit Proteasome. eLife 2020, 9, e56825.
- Stykel, M.G.; Humphries, K.M.; Kamski-Hennekam, E.; Buchner-Duby, B.; Porte-Trachsel, N.; Ryan, T.; Coackley, C.L.; Bamm, V.V.; Harauz, G.; Ryan, S.D. α-Synuclein Mutation Impairs Processing of Endomembrane Compartments and Promotes Exocytosis and Seeding of α-Synuclein Pathology. Cell Rep. 2021, 35, 109099.
- Leitão, A.D.G.; Rudolffi-Soto, P.; Chappard, A.; Bhumkar, A.; Lau, D.; Hunter, D.J.B.; Gambin, Y.; Sierecki, E. Selectivity of Lewy Body Protein Interactions along the Aggregation Pathway of α-Synuclein. Commun. Biol. 2021, 4, 1124.
- Joshi, N.; Raveendran, A.; Nagotu, S. Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases 2020, 8, 24.
- Ellis, C.E.; Schwartzberg, P.L.; Grider, T.L.; Fink, D.W.; Nussbaum, R.L. A-Synuclein Is Phosphorylated by Members of the Src Family of Protein-Tyrosine Kinases *. J. Biol. Chem. 2001, 276, 3879–3884.
- Arawaka, S.; Wada, M.; Goto, S.; Karube, H.; Sakamoto, M.; Ren, C.H.; Koyama, S.; Nagasawa, H.; Kimura, H.; Kawanami, T.; et al. The Role of G-Protein-Coupled Receptor Kinase 5 in Pathogenesis of Sporadic Parkinson’s Disease. J. Neurosci. 2006, 26, 9227–9238.
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. C-Abl Phosphorylates α-Synuclein and Regulates Its Degradation: Implication for α-Synuclein Clearance and Contribution to the Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet. 2014, 23, 2858–2879.
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of α-Synuclein by Chaperones in Mammalian Cells. Nature 2020, 577, 127–132.
- Wegmann, S.; Biernat, J.; Mandelkow, E. A Current View on Tau Protein Phosphorylation in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2021, 69, 131–138.
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of Tau and Alpha-Synuclein Epitopes in Lewy Bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397.
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the Synucleinopathies: Alpha Synuclein as a Driving Force in Neurodegenerative Comorbidities. Transl. Neurodegener. 2019, 8, 28.
- Waxman, E.A.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by α-Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618.
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103.
- Pan, L.; Li, C.; Meng, L.; Tian, Y.; He, M.; Yuan, X.; Zhang, G.; Zhang, Z.; Xiong, J.; Chen, G.; et al. Tau Accelerates α-Synuclein Aggregation and Spreading in Parkinson’s Disease. Brain 2022, 145, 3454–3471.
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J. Neurosci. 2017, 37, 11406–11423.
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular Milieu Imparts Distinct Pathological α-Synuclein Strains in α-Synucleinopathies. Nature 2018, 557, 558–563.
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469.
- Rey, N.L.; Bousset, L.; George, S.; Madaj, Z.; Meyerdirk, L.; Schulz, E.; Steiner, J.A.; Melki, R.; Brundin, P. α-Synuclein Conformational Strains Spread, Seed and Target Neuronal Cells Differentially after Injection into the Olfactory Bulb. Acta Neuropathol. Commun. 2019, 7, 221.
- Leitao, A.; Bhumkar, A.; Hunter, D.J.B.; Gambin, Y.; Sierecki, E. Unveiling a Selective Mechanism for the Inhibition of α-Synuclein Aggregation by β-Synuclein. Int. J. Mol. Sci. 2018, 19, 334.
- Bisi, N.; Feni, L.; Peqini, K.; Pérez-Peña, H.; Ongeri, S.; Pieraccini, S.; Pellegrino, S. α-Synuclein: An All-Inclusive Trip Around Its Structure, Influencing Factors and Applied Techniques. Front. Chem. 2021, 9, 666585.
- Song, Y.J.C.; Lundvig, D.M.S.; Huang, Y.; Wei, P.G.; Blumbergs, P.C.; Højrup, P.; Otzen, D.; Halliday, G.M.; Jensen, P.H. P25α Relocalizes in Oligodendroglia from Myelin to Cytoplasmic Inclusions in Multiple System Atrophy. Am. J. Pathol. 2007, 171, 1291–1303.
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple System Atrophy-Associated Oligodendroglial Protein P25α Stimulates Formation of Novel α-Synuclein Strain with Enhanced Neurodegenerative Potential. Acta Neuropathol. 2021, 142, 87–115.
- Woerman, A.L.; Oehler, A.; Kazmi, S.A.; Lee, J.; Halliday, G.M.; Middleton, L.T.; Gentleman, S.M.; Mordes, D.A.; Spina, S.; Grinberg, L.T.; et al. Multiple System Atrophy Prions Retain Strain Specificity after Serial Propagation in Two Different Tg(SNCA*A53T) Mouse Lines. Acta Neuropathol. 2019, 137, 437–454.
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol. 2020, 139, 977–1000.
- Holec, S.A.M.; Woerman, A.L. Evidence of Distinct α-Synuclein Strains Underlying Disease Heterogeneity. Acta Neuropathol. 2021, 142, 73–86.
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s Disease The Dual Hit Theory Revisited. Ann. N. Y. Acad. Sci. 2009, 1170, 615–622.
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-Synuclein Preformed Fibrils into the Mouse Gastrointestinal Tract Induces Lewy Body-like Aggregates in the Brainstem via the Vagus Nerve. Mol. Neurodegener. 2018, 13, 21.
- Gray, M.T.; Munoz, D.G.; Schlossmacher, M.G.; Gray, D.A.; Woulfe, J.M. Protective Effect of Vagotomy Suggests Source Organ for Parkinson Disease. Ann. Neurol. 2015, 78, 834–835.
- Mendes, A.; Gonçalves, A.; Vila-Chã, N.; Moreira, I.; Fernandes, J.; Damásio, J.; Teixeira-Pinto, A.; Taipa, R.; Lima, A.B.; Cavaco, S. Appendectomy May Delay Parkinson’s Disease Onset. J. Mov. Disord. 2015, 30, 1404–1407.
- Lu, H.-T.; Shen, Q.-Y.; Xie, D.; Zhao, Q.-Z.; Xu, Y.-M. Lack of Association between Appendectomy and Parkinson’s Disease: A Systematic Review and Meta-Analysis. Aging Clin. Exp. Res. 2020, 32, 2201–2209.
- Borghammer, P.; Hamani, C. Preventing Parkinson Disease by Vagotomy Fact or Fiction? Neurology 2017, 88, 1982.
- Peelaerts, W.; Bousset, L.; der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344.
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-Seeded α-Synuclein Fibrils Promote Gut Dysfunction and Brain Pathology Specifically in Aged Mice. Nat. Neurosci. 2020, 23, 327–336.
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for Bidirectional and Trans-Synaptic Parasympathetic and Sympathetic Propagation of Alpha-Synuclein in Rats. Acta Neuropathol. 2019, 138, 535–550.
- Veys, L.; Van Houcke, J.; Aerts, J.; Van Pottelberge, S.; Mahieu, M.; Coens, A.; Melki, R.; Moechars, D.; De Muynck, L.; De Groef, L. Absence of Uptake and Prion-Like Spreading of Alpha-Synuclein and Tau after Intravitreal Injection of Preformed Fibrils. Front. Aging Neurosci. 2021, 12, 614587.
- Ding, X.; Zhou, L.; Jiang, X.; Liu, H.; Yao, J.; Zhang, R.; Liang, D.; Wang, F.; Ma, M.; Tang, B.; et al. Propagation of Pathological α-Synuclein from the Urogenital Tract to the Brain Initiates MSA-like Syndrome. iScience 2020, 23, 101166.
- Just, M.K.; Gram, H.; Theologidis, V.; Jensen, P.H.; Nilsson, K.P.R.; Lindgren, M.; Knudsen, K.; Borghammer, P.; Van Den Berge, N. Alpha-Synuclein Strain Variability in Body-First and Brain-First Synucleinopathies. Front. Aging Neurosci. 2022, 14, 907293.

