Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | María Laura Ruiz | -- | 6875 | 2023-08-01 19:32:08 | | | |
| 2 | Rita Xu | -7 word(s) | 6868 | 2023-08-02 07:25:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bucci-Muñoz, M.; Gola, A.M.; Rigalli, J.P.; Ceballos, M.P.; Ruiz, M.L. Extracellular Vesicles and Cancer Multidrug Resistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/47508 (accessed on 14 June 2026).
Bucci-Muñoz M, Gola AM, Rigalli JP, Ceballos MP, Ruiz ML. Extracellular Vesicles and Cancer Multidrug Resistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/47508. Accessed June 14, 2026.
Bucci-Muñoz, María, Aldana Magalí Gola, Juan Pablo Rigalli, María Paula Ceballos, María Laura Ruiz. "Extracellular Vesicles and Cancer Multidrug Resistance" Encyclopedia, https://encyclopedia.pub/entry/47508 (accessed June 14, 2026).
Bucci-Muñoz, M., Gola, A.M., Rigalli, J.P., Ceballos, M.P., & Ruiz, M.L. (2023, August 01). Extracellular Vesicles and Cancer Multidrug Resistance. In Encyclopedia. https://encyclopedia.pub/entry/47508
Bucci-Muñoz, María, et al. "Extracellular Vesicles and Cancer Multidrug Resistance." Encyclopedia. Web. 01 August, 2023.
Copy Citation
Cancer multidrug resistance (MDR) is one of the main mechanisms contributing to therapy failure and mortality. Overexpression of drug transporters of the ABC family (ATP-binding cassette) is a major cause of MDR. Extracellular vesicles (EVs) are nanoparticles released by most cells of the organism involved in cell–cell communication. Their cargo mainly comprises, proteins, nucleic acids, and lipids, which are transferred from a donor cell to a target cell and lead to phenotypical changes.
resistance protein
multidrug resistance-associated protein
extracellular vesicles
1. Introduction
1.1. Multidrug Resistance (MDR)
Around the world, cancer ranks as a leading cause of death and represents a significant barrier to raising life expectancy [1]. According to the estimates performed by GLOBOCAN in 2020, the total number of new cancer cases and deaths was recorded as 19.3 million and almost 10.0 million worldwide, respectively [2]. The most commonly diagnosed cancer is female breast cancer (2.3 million new cases; 11.7%), followed by lung (11.4%), colorectal (10.0%), prostate (7.3%), and stomach (5.6%) cancers. Regarding mortality, lung cancer is the leading cause of cancer death (1.8 million deaths; 18%), followed by colorectal (9.4%), liver (8.3%), stomach (7.7%), and female breast (6.9%) cancers. The global cancer burden is expected to increase by 47% by the year 2040, reaching 28.4 million cases [2].
In order to improve the lifespan of cancer patients and to eradicate cancer, several therapeutic strategies have been explored, from highly invasive surgical resections to radiation-induced eradication and diverse therapeutic approaches like chemotherapy, targeted therapy, and, more recently, immunotherapy [3][4]. However, several factors like the age of diagnosis, treatment time, and stage of cancer, among others, affect the efficacy of treatment. In addition, most drugs present serious adverse effects due to the lack of target selectivity [3]. More importantly, resistance to already available drugs, which allows cancer to proliferate in the presence of a chemotherapeutic agent, has emerged as a major obstacle for cancer therapies, being associated with more than 90% of cases of treatment failure [3][5]. This mechanism, known as multidrug resistance (MDR) or chemoresistance, consists of the resistance of tumor cells to different drugs with multiple chemical structures, multiple mechanisms of action, and multiple targets and, ultimately, renders the cancer cell ineffective in achieving the therapeutically required intracellular drug concentration. Indeed, cancer cells present cross-resistance to a broad spectrum of structurally unrelated chemotherapeutic agents and this mechanism of MDR can be intrinsic or acquired. Intrinsic resistance refers to a complete failure of response to initial antitumor therapy in patients, i.e., cancer cells are already resistant before treatment. In acquired resistance, on the other hand, cancer cells become resistant during treatment and therefore the cancer initially responds to therapy but then relapses or progresses after a period of time. Most tumors in clinical settings become resistant owing to a combination of both types of resistance [3][5]. Unfortunately, the mechanisms implicated are not yet fully understood, and MDR is the major cause of cancer recurrence and metastasis, and it is related to a longer period of hospitalization, increased medical costs, and high mortality [4].
Frequently, when tumor cells face a new stress, such as a chemotherapeutic agent, they may adapt by acquiring a series of mutations, become resistant and, in this way, survive therapy [6]. MDR is a multifactorial phenomenon, with diverse resistance mechanisms to the same chemotherapeutic agent acting on the same cancer cell. In this way, it is very complex to inhibit MDR because when one resistance mechanism is blocked, cancer cells adapt and develop MDR through other mechanisms. The multiple mechanisms, which are associated with a complex process of multiple genes, factors, pathways, and multiple steps, may include host factors, tumor factors, as well as tumor–host interactions [6][7]; microbiomes may also play a role in cancer drug resistance [8].
Host factors include the genetic variants between patients that affect the efficacy of therapy, like single nucleotide variants, insertions, deletions, repeats, chromosomal rearrangement, and copy number variations [3][6]. Also, cancer patients are very susceptible to drug–drug interactions because they commonly receive complementary medications during therapy. Coadministered drugs may change the efficacy and/or toxicity of the chemotherapeutic agent by modifying its absorption, distribution, metabolism, or excretion (pharmacokinetic) and can result in synergistic, antagonistic, or additive responses (pharmacodynamics) [7].
Several tumor factors have been described and, additionally, the function of some of the proteins involved in these mechanisms could be affected by the presence of genetic variants between individuals (host factors) [4][7][9]. The predominant mechanism contributing to MDR results from the increased drug efflux by ATP-dependent pumps located on the surface of cancer cells. These transmembrane transporters belong to the ATP-binding cassette (ABC) superfamily and their expression is frequently upregulated in tumor cells [3][4][5]. Also, drug efflux can be mediated by extracellular vesicles (EVs) with the ability to sequester chemotherapeutic drugs, extruding them from cells once released from tumor cells. ABC transporters have been found in the cargo of EVs in an inverted orientation, contributing to the influx of drugs into the EVs [7]. Similarly, another mechanism is the reduced uptake of the drug due to changes in the expression and/or activity of transporters involved in the transport of the chemotherapeutic agent inside the cancer cell or by the reduction in drug diffusion by a barrier to drug permeability established in these cells. The sequestration of chemotherapeutic agents in intracellular vesicles and compartments away from the cellular target, like the lysosomal compartmentalization of drugs, also contributes to MDR. In addition, alterations in the biotransformation or metabolism of the drug by modifications in the expression and/or activity of drug-metabolizing enzymes could lead to impaired prodrug activation or increased proportion of inactive metabolites, reducing the intracellular concentration of the active drug [3][7][9]. Since drugs need to be active and reach their targets at an adequate concentration to achieve therapeutic efficacy, the above mechanisms directly influence the outcome of therapy by decreasing the available intracellular drug concentration [9].
The plethora of tumor factors also involve mutations or changes in the expression levels of target genes of the antitumor drugs that prevent an efficient drug–target interaction in proteins involved in survival and cell death (that allow cancer cells to survive) and in proteins related to DNA repair mechanisms (which enable cancer cells to repair the DNA damaged by chemotherapeutic agents and avoid cellular death while promoting genomic instability and mutations) [3][4][9]. Epigenetic alterations, such as DNA methylation and histone and chromatin modifications, are another tumor factor that can change the expression of numerous MDR genes, like those coding for ABC transporters. Other tumor factors are the deregulation of microRNA (miRNAs) implicated in the regulation of MDR proteins, modifications in the epithelial–mesenchymal transition (which leads to a dedifferentiation of epithelial cells to a mesenchymal phenotype), and cancer stemness related to cancer stem cells. Finally, the heterogeneity within an individual tumor or between tumors (interindividual) is another tumor factor that provides genetic and epigenetic diversity, allowing for therapy-induced expansion of preexisting drug-resistant cancer cell clones [4][6][9].
Tumor–host interactions are mainly linked to the tumor microenvironment (TME), a highly complex ecosystem consisting of extracellular matrix, soluble factors, and tumor and stromal cells, which imposes a selective pressure that drives tumor progression [3][7]. This selective pressure includes hypoxia, extracellular acidosis, nutrient deprivation, immune surveillance, and unorganized leaky vasculature, which influence MDR by altering the distribution of drugs inside tumors and also by affecting their cellular uptake. In addition, some of these characteristics of the TME modify the expression of MDR proteins, such as the ABC transporters. In addition to the aforementioned MDR mechanisms, other specific ones linked to the immune cells infiltrating the TME have also been described [4][10]. Additionally, extracellular vesicles (EVs) might contribute to the modulation of TME and to the dissemination of MDR by transferring MDR-associated proteins between tumor and stromal cells and between drug-resistant and drug-sensitive tumor cells [7].
The TME does not only bring complexity to the MDR phenomenon due to its diverse effects on chemoresistance, but also to the effect of microbes that can translocate and reside in tumor niches. The microbes are able to inactivate anticancer drugs via biotransformation or by activating oncogenic pathways inducing chemoresistance. In patients with colorectal cancer, an important intratumoral bacteria, Fusobacterium nucleatum, has been shown to promote the development of oxaliplatin resistance during treatment by activating the innate immune system and inducing autophagy [8]. Increased levels of the oral pathogens Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis were observed in pancreatic cancer patients, which promoted the expression of cytidine deaminase and impacted the occurrence of chemoresistance. In the mouse colorectal cancer model CT26, intratumoral administration of E. coli not only affected the activity and concentration of gemcitabine at the tumor site, but also affected the development of drug resistance [11][12]. Intratumoral gammaproteobacteria with long isoform of cytidine deaminase can inactivate gemcitabine, leading to chemoresistance [12][13]. Intratumor-residing gut microbiota could modulate chemokine levels and affect CD8+ T cell infiltration, consequently influencing patient survival in cutaneous melanoma. Manipulating the intratumor gut microbiome may benefit patient outcomes for those with immunotherapy [14].
All mechanisms mentioned in this section are illustrated in Figure 1.
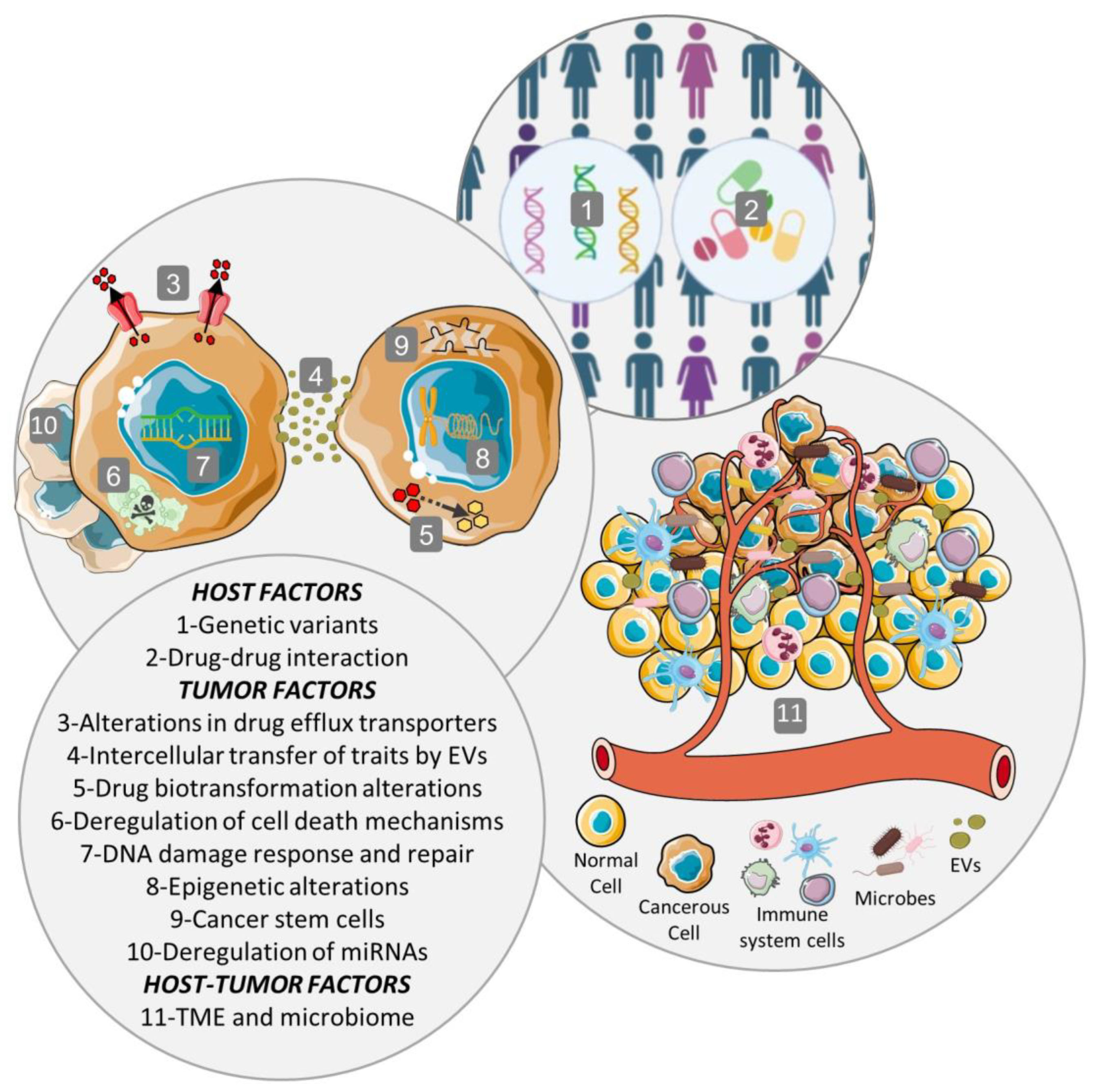
Figure 1. Multifactorial phenomenon of MDR. The multiple mechanisms, which are associated with a complex process of multiple genes, factors, pathways, and multiple steps, may include host factors, tumor factors, as well as tumor–host interactions. Host factors include host genetic variants and drug–drug interactions. Tumor factors comprise alterations in intracellular drug concentration originated by modifications in drug efflux or influx transporters or by extracellular vesicles (EVs), alterations in the biotransformation or metabolism of the drug, deregulation of cell death mechanisms, DNA damage response and repair, epigenetic alterations, cancer stem cells, and deregulation of microRNAs (miRNAs). Among the tumor–host factors are the tumor microenvironment (TME), the selective pressures and tumor evolution, intercellular transfer of traits by extracellular vesicles (EVs), and tumor microbiome.
1.2. ABC Transporters
The ABC superfamily is identified by the presence of a group of highly conserved motifs. Structurally, all ABC transporters consist of at least two transmembrane domains (TMDs), which are inserted in the lipid bilayer, and two nucleotide-binding domains (NBDs) located in the cytoplasm [15]. Some exceptions to this rule are transporters whose sequence spans a single NBD and a single TMD. In the latter case, the transporter needs to be dimerized to obtain full functionality [16]. TMDs have a conserved three-dimensional structure, consisting of a core of six transmembrane α-helices per TMD. These domains are hydrophobic and structurally diverse; they recognize and translocate various substrates through conformational changes. The variability of the sequences and structures of the different TMDs is reflected in the chemical diversity of the substrates they transport [17]. NBDs are domains consisting of a highly conserved ABC motif, which is responsible for binding and hydrolyzing ATP, thus providing the energy for the transport of physiological and xenobiotic substrates [18]. In addition to the ABC motif, NBDs also contain other motifs and typical conformations that characterize them, such as the Walker A and B motifs [19].
1.2.1. P-Glycoprotein/ABCB1/MDR1
The first member of the ABC transporter superfamily, P-gp (ABCB1/MDR1), was identified in 1976 by Juliano and Ling as a 170 kDa membrane glycoprotein. P-gp was found overexpressed in colchicine-resistant ovarian cell lines and reduced the permeability to this drug [20]. After that, it became the most studied ABC transporter and the one with the broadest substrate specificity [21], as it transports a large variety of molecules with different chemical structures and molecular weights [22]. There is a positive correlation between MDR1 expression and high risk of disease progression in ovarian cancer patients, as was demonstrated previously [23]. Additionally, an inverse relationship between paclitaxel response and P-gp expression has been reported [24].
In epithelial polarized cells, P-gp localizes to the apical membrane of renal proximal tubular cells, cells from placenta, hepatocytes, enterocytes, adrenal cells, and blood–brain barrier cells, where it functions to protect against xenobiotics and cellular toxicants [25][26][27][28][29]. P-gp is also expressed on CD34+ hematopoietic progenitor cells, natural killer cells, and CD8+ T cells [30]. P-gp expression was analyzed in 60 cell lines from a wide range of tumors and detected in 39 of them using quantitative real-time PCR. The highest levels of P-gp were found in renal and colon carcinomas [31]. P-gp is frequently also detected in cancer stem-like cells.
MDR1 (i.e, gene codifying P-gp) expression can be induced by a variety of environmental stimuli, such as ultraviolet radiation [32]. MDR1 expression is also induced by compounds such as sodium butyrate, retinoic acid, phorbol esters, and even many chemotherapeutic drugs [32]. The latter are able to induce specific epigenetic modifications at the MDR1 locus, together with the induction of P-gp, at protein level, through transcriptional activation. Such a mechanism is dependent on the methylation status of the MDR1 promoter [33]. Binding of specific transcription factors to DNA sequences, located distal or proximal to the MDR1 promoter region, as well as methylation/demethylation, modulate MDR1 transcriptional expression [34]. Hypermethylation of the MDR1 promoter has been found to be associated with activation of transcription-blocking mechanisms, which are independent of histone deacetylase. Methylation and acetylation of lysine residues in the N-terminal fragments of histones are important mechanisms of MDR1 expression regulation [35]. Scotto and colleagues (2003) have shown that the previously mentioned stimuli, inducers of MDR1 expression, converge on a region of the promoter of this gene, which they called the “enhancer”. This promoter region, like that of most genes that do not have a “TATA box” in their sequence, includes an inverted “CCAAT box” (which interacts, for example, with the trimeric transcription factor NF-Y) and a GC-rich element (which interacts with members of the Sp family of transcription factors, such as Sp1) [34]. In addition to NF-Y and Sp1, P-gp expression is also transcriptionally regulated by nuclear receptors that act as xenosensors. Among them researchers can name the pregnane X receptor (PXR) and the constitutive androstane receptor (CAR), which induce MDR1 transcription [36][37]. Under oxidative stress conditions, there is evidence that the nuclear factor erythroid 2-related factor 2 (Nrf2), which is activated to protect the cell from free radical damage, is able to modulate MDR1 expression [38][39]. On the other hand, several steroid hormones can also modulate MDR1 through the estrogen receptor (ER, estrogen receptor) [40][41]. MDR1 promoter activity can also be stimulated by the tumor suppressor protein p53 and the Ras oncogene product proteins Ras, c-Raf, and c-Raf kinase [5]. P-gp expression can also be regulated at the posttranscriptional level. MiRNAs are a class of short, noncoding RNA molecules that regulate gene expression. Through bioinformatics analysis, several miRNAs were found to bind to the 3’ end of the untranslated regions (UTRs) of the MDR1 mRNA [5]. In fact, mRNA UTRs can play an essential role in regulating protein synthesis and the half-life of different mRNAs in the cell. For example, the miRNAs miR-223 and miR-145 decrease P-gp expression by direct action on the 3’-UTR of MDR1 mRNA, binding to it and preventing translation. miRNA-27a and miRNA-138, instead, regulate P-gp expression at the level of transcription [42]. In addition to miRNAs, several drugs, such as colchicine, doxorubicin, colcemid, vinblastine, cytarabine, and ivermectin, regulate P-gp expression by stabilizing its mRNA [5][35][43][44].
1.2.2. MRPs/ABCCs
The ABCC/MRP subfamily consists of at least nine homologs expressed in humans, named MRP1 to MRP9 [45][46][47][48]. They exhibit a conserved structure but heterogeneity in terms of substrate specificity, localization, and function. The first member of this subfamily, MRP1, was discovered by Cole in 1992, in a lung cancer cell line resistant to doxorubicin without overexpression of P-gp [49].
MRP1 is expressed in normal tissues such as the kidney, intestine, blood–brain barrier, lung, testis, and mononuclear cells [50]. Normal liver expresses MRP1 at very low levels [51]. However, an isoform of MRP1, located in the canalicular membrane, was later discovered in the liver and named MRP2 [52]. Its expression is not restricted only to the liver, but is also present in the kidney, small intestine, and blood–brain barrier [53]. Subsequently, a basolateral isoform called MRP3/ABCC3 [54] was identified in the sinusoidal membrane of hepatocytes, where it mediates the vectorial transport of substrates from the hepatocyte to the sinusoidal blood [54][55][56]. It was also detected in the kidney, intestine, pancreas, lung, gallbladder [57][58], as well as in different tumors where its expression was associated with increased chemoresistance [59][60]. MRP1 is capable of transporting chemotherapeutic agents such as anthracyclines, camptothecin analogues, and vinca alkaloids. MRP1 is overexpressed in lung, breast, prostate, and ovarian cancer as well as in gastrointestinal carcinoma, melanoma, and leukemia [61]. MRP2 is capable of transporting conjugated compounds like acetaminophen glucuronide or ethynyl estradiol glucuronide [53] and also unconjugated drugs such as methotrexate, ritonavir, saquinavir, vinblastine, sorafenib, and cisplatin [53][62]. MRP3 is able to transport and thus participate in methotrexate and etoposide resistance [59][60]. MRP3 has also been shown to participate in resistance to sorafenib in HCC [63]. Additionally, MRP4 and -5 can also confer resistance against some chemotherapeutic drugs and are able to transport certain organic anions [64][65][66].
1.2.3. BCRP/ABCG2
In 1998, Doyle et al. described an ATP-dependent reduction in the intracellular accumulation of anthracyclines in MCF7 cells and resistance to doxorubicin, without overexpression of known multidrug resistance transporters such as P-gp or MRPs [67]. This new ABC transporter was named BCRP (breast cancer-resistant protein/ABCG2). The human BCRP gene is located on chromosome 4q22 4q21, and encodes a unique 72 kDa peptide consisting of only one NBD and one TMD, and is therefore referred to as a “half transporter” [68]. BCRP is also known as MXR, because it mediates the efflux of mitoxantrone and is therefore responsible for resistance to this frequently used chemotherapeutic compound [69] in tumor cells. In addition, this transporter is associated with drug resistance in breast cancer [70]. BCRP is expressed in numerous organs/tissues, including the placenta, liver, gastrointestinal tract, kidney, prostate, breast, adrenal gland, and the luminal surface of endothelial cells of human brain microvessels. BCRP protects normal cells from xenobiotic toxicity in order to maintain physiological homeostasis [71]. Overexpression of BCRP renders cancer cells resistant to multiple drugs such as mitoxantrone, topotecan, and methotrexate and is associated with poor response to chemotherapy in leukemia and breast cancer patients [61]. In addition, a negative correlation between BCRP expression and prognosis in breast cancer has been described [72].
1.3. Extracellular Vesicles
Extracellular vesicles (EVs) are particles released by most cells of the organism to the extracellular space. According to their mechanisms of biogenesis and release, EVs are classified into exosomes, microvesicles (MVs, also known as ectosomes or microparticles), and apoptotic bodies. Exosomes originate from endosomal compartments. MVs originate through outward budding of the plasma membrane. Apoptotic bodies are released during programmed cell death. In terms of their size, most exosomes have a diameter between 40 and 100 nm, MV diameters usually fall between 200 and 1000 nm, and apoptotic bodies are larger than 1 µm. The EV cargo comprises proteins, nucleic acids (mRNAs, miRNAs, other noncoding RNAs), carbohydrates, and lipids. Although size distributions partially allow for distinguishing between exosomes, MVs, and apoptotic bodies, from smaller to larger particle sizes, particle distributions also overlap [73]. In addition, the current EV isolation methods (e.g., ultracentrifugation, size exclusion chromatography, precipitation, ultrafiltration, affinity capture) are mostly based on physicochemical properties common to more than one type of EV. Therefore, the isolation of a specific type of vesicles remains a difficult, mostly unaccomplished task. In agreement with the recommendations of the International Society for Extracellular Vesicles (ISEV) [74], researchers use the term EVs throughout the manuscript, irrespective of the nomenclature used in the original sources. A clear mention to a specific EV type (e.g., MVs) is made only when the isolation methods used clearly allow for the isolation of that type of EV with negligible contamination by other types of vesicle.
Functionally, EVs have been demonstrated to play a major role in cell–cell communication in health [75] and disease [76]. EV participation in the pathogenesis of several disorders has been demonstrated. Among them, EVs have been reported to play a major role in cancer cell proliferation, metastasis, and immune evasion [76]. Several studies have also addressed the role of EVs in relation with cancer multidrug resistance using different models in vitro and in vivo.
2. EVs as Mediators of Multidrug Resistance
The most frequent mechanism of EV-mediated drug resistance consists of the release of EVs by donor cells with a MDR phenotype, followed by uptake by drug-sensitive cells (i.e., target cells), which, in response, acquire a drug-resistant phenotype. Furthermore, donor cells may not necessarily be tumor- or multidrug-resistant cells. For instance, canine natural killer cell-derived EVs injected to a mouse xenograft bearing canine mammary tumors resulted in an increase in P-gp expression in the tumor cells. Although the functional relevance of P-gp upregulation was not investigated, this research clearly highlights the wide spectrum of EV-mediated cell–cell interactions with a potential impact on transporter modulation and, most likely, drug resistance [77].
Cell–cell communication via EVs is strongly determined by the cargo transported between donor and target cells. Furthermore, cargo-independent features may also influence the transfer of information. For instance, a study with drug-sensitive and multidrug-resistant non-small cell lung cancer (NCI-H460) and chronic myeloid leukemia (K562) cells identified an increase in EV release by drug-resistant cells. This observation could be attributed to the upregulation of the Rab GTPases Rab5 and Rab11, and Rab 27 in drug-resistant NCI-H460 and K562 cells, respectively, compared to their drug-sensitive counterparts. In addition, drug-sensitive cells exhibited an increase in EV uptake, independent of the phenotype of the donor cells. These features, although not directly impacting the EV cargo, may also favor the vesicle-mediated cell–cell communication process [78]. Another study investigated the size distribution of EVs released by the same cell types. Results pointed to a predominant release of larger EVs by drug-resistant NCI-H460 and K562 cells. On the contrary, drug-sensitive cells release a higher proportion of exosomes, as determined based on the EV size and the expression of exosome markers [79].
A similar study, using drug-sensitive and drug-resistant cervical cancer cells, found higher P-gp expression in EVs released by the resistant clones. However, incubation of nontumoral cells (i.e., immortalized human fibroblasts) with EVs did not result in a transfer of the transporter or in the stimulation of other malignant features (i.e., migration) in target cells [80]. These findings point to additional cell-specific mechanisms regulating and influencing the directional transfer of information between different cell types. Thus, although the actual cargo of the EVs is the main factor determining the effect in terms of multidrug resistance, the identity as well as structural and functional features of donor and target cells may play a relevant role in the transfer of the drug-resistant phenotype as well. Researchers discuss different cases of EV-mediated multidrug resistance based on the role played by vesicles and bioactive cargo. The most common mechanisms are also schematized in Figure 2. In addition, researchers address the potential of EVs as a source of biomarkers of drug-resistance.
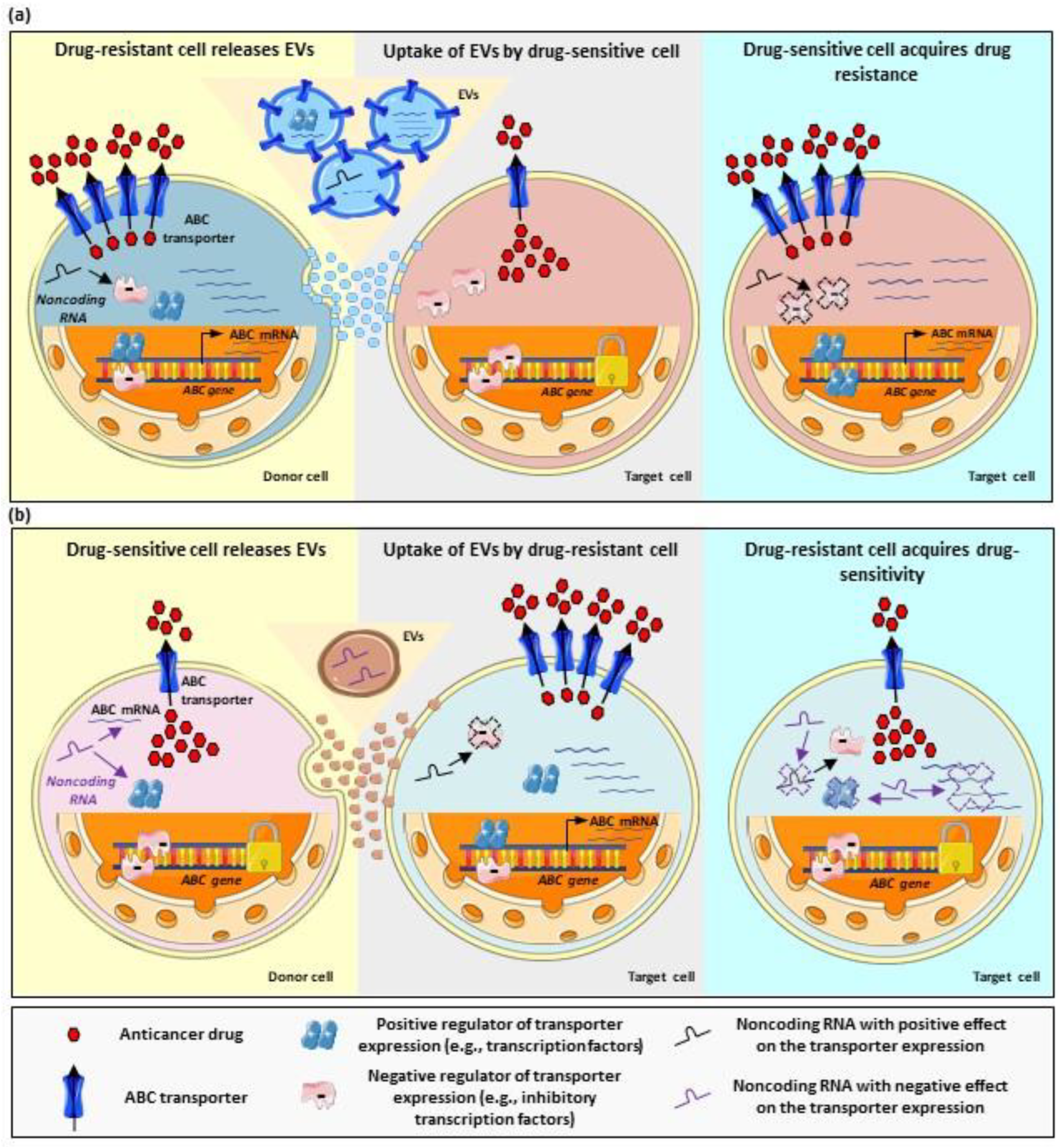
Figure 2. Regulation of multidrug resistance by EV-mediated cell–cell communication. (a) Stimulation of drug resistance by EVs. Drug-resistant cells (donor cells) with overexpression of ABC transporters exhibit lower accumulation of anticancer drugs (left panel). These cells can release EVs carrying, for example, ABC transporters, their coding mRNAs, noncoding RNAs, and proteins with a positive regulatory function. Once taken up by target cells (originally drug-sensitive cells, central panel), the components of the EV cargo result in the upregulation of ABC transporters. This takes place, for example, by direct shuttling of transporter molecules or their codifying mRNAs; but also by shuttling of regulatory noncoding RNAs, which may inhibit the function of negative regulators; or by shuttling of positive regulators (e.g., transcription factors). Ultimately, the stimulation of drug resistance in the target cell takes place (right panel). (b) Inhibition of drug resistance by EVs. Drug-sensitive cells (donor cells, left panel) exhibit lower transporter expression and, therefore, increased drug accumulation. These cells usually overexpress negative regulators of the transporter expression and the function of positive regulators may be inhibited (for example, by noncoding RNAs). These cells release EVs, which can be taken up by target cells, originally drug-resistant (central panel), with transporter overexpression. The transfer of the EV cargo to target cells results in transporter downregulation, for example, by interaction of noncoding RNAs with the transporter mRNA and, therefore, inhibition of the translation; by inhibition of positive regulatory factors; or by activation of negative regulatory factors. Arrows indicate situations of up-regulation, down-regulation or inhibition.
2.1. EV-Mediated Shuttle of ABC Transporters
One of the most frequently described mechanisms of EV-mediated drug resistance is the direct transfer of drug transporters or its coding mRNA from a donor, in general, drug-resistant cell to a target, mostly, drug-sensitive cell. In this regard, using a coculture system, Pasquier et al. demonstrated the transfer of P-gp from drug-resistant to drug-sensitive MCF7 cells via EVs. As a result of this interaction, target cells acquired doxorubicin resistance. The authors also demonstrated higher P-gp levels in the vesicles derived from resistant cells compared to the vesicles obtained from drug-sensitive cells. Since both cell types were separated by a filter with a 0.4 μm pore size, EVs in the lower size range are likely to be involved in the transfer of resistance. Interestingly, the time dependence of the transfer was also investigated. Increased P-gp protein expression in target cells was already observed at 4 h of exposure to the EVs. Peak expression was registered at 12 h of exposure and a return to basal levels was observed from 36 h of exposure in advance. Additionally, an increase in P-gp activity in target cells was observed. Here, the kinetics of the effect exhibited a similar pattern, with the highest increase in transporter activity observed at 4 h of exposure and a return to basal levels at 24 h of exposure to the EVs [81]. Using a similar experimental setting, Wang et al. demonstrated the upregulation of P-gp and MRP1 mRNA in sensitive MCF7 cells treated with EVs from a doxorubicin-resistant MCF7 clone. As expected, treatment with EVs also stimulated resistance to doxorubicin in the sensitive clone. Higher P-gp protein levels in EVs from the resistant clone were observed. However, no data on the mRNA expression in the vesicles were obtained. Therefore, it is still unclear whether the mRNA upregulation mediated by EV treatment is a mere result of messenger transfer via EVs or whether it results from the transfer of other regulatory proteins or RNAs. Interestingly, in this study, the authors demonstrated an inhibitory effect of psoralen, a furanocoumarin, on EV release by the doxorubicin-resistant cells, thus suggesting a potential compound able to counteract the transfer of drug resistance [82]. Similar findings were described in another study using docetaxel-resistant MCF7 cells due to P-gp overexpression. As expected, EVs derived from these cells exhibited a higher expression of P-gp compared to those from drug-sensitive MCF7 cells and, once added to the latter, resulted in an increase in P-gp protein levels and resistance to docetaxel in target cells [83].
Another study using MCF7 and CEM (human acute lymphoblastic leukemia) cells investigated P-gp in MVs (300–600 nm) released by drug-resistant clones of both cell types. Addition of these MVs to drug-sensitive clones of the respective cell line resulted in significant P-gp upregulation at the mRNA level. Unexpectedly, a simultaneous decrease in MRP1 mRNA expression was observed in CEM cells treated with MVs from the resistant clone. The authors postulated that miR-326, which negatively regulates MRP1 mRNA expression and is carried in EVs together with transporter messengers, was responsible for MRP1 downregulation. These findings point to a complex picture of EV-mediated transfer of ABC transporters, where specific transporters can be up- or downregulated based on the cargo of the EVs. Concomitantly, increased or decreased resistance to specific chemotherapeutic agents, could be expected [84]. A further study from the same group analyzed, in addition, the cell specificity of transporter transfer via MVs. Interestingly, the nature of the donor cells was reported to play a key role. In fact, MVs derived from drug-resistant CEM cells transferred P-gp and MRP1 to both cancerous and noncancerous cells (i.e., human mammary basal epithelial cells, human osteoblasts, and human urothelial cells) while, on the contrary, MVs from drug-resistant MCF7 cells only transferred P-gp to cancerous cells (i.e., drug-sensitive MCF7). The presence of CD44 on the surface of EVs derived from resistant MCF7 cells and the absence of this surface marker on the EVs from resistant CEM cells was proposed as a possible explanation for this cell-specific transfer [85]. The pathophysiological relevance of transporter transfer to nontumoral cells still has to be elucidated. Furthermore, it should be noted that the set of nonmalignant cells used for this research was rather arbitrarily selected. It would be interesting, for instance, to investigate the effect of tumoral EVs on nontumoral cells in the tumor microenviroment, as the latter may also influence drug resistance [86] and, ultimately, cancer prognosis.
Transfer of drug resistance via MVs in human acute lymphoblastic leukemia was also reported in another study using a similar model. In this case, P-gp-containing MVs were isolated from drug-resistant CEM cells and added to a culture of drug-sensitive cells. As expected, an increase in P-gp protein expression in target cells was observed. Furthermore, target cells exhibited increased efflux of the P-gp substrate rhodamine 123 [87]. Transfer of functional P-gp was also observed in KB cells, originally classified as human oral epidermoid carcinoma [88]. Here, a vincristine-resistant clone was reported to express higher levels of P-gp. Furthermore, vincristine triggered a concentration-dependent increase in the P-gp content secreted in the EVs. In addition, using a coculture system in transwells, the authors demonstrated the transfer of functional P-gp from drug-resistant to drug-sensitive cells. Noteworthy, no P-gp mRNA was detected in target cells, even at high concentrations of EVs, thus supporting the transfer of P-gp only at the protein level from donor (resistant) to target (sensitive) cells. In addition, EVs from donor cells enhanced the resistance of target cells to doxorubicin, a well-known P-gp substrate, this process being prevented by incubation with the P-gp inhibitor verapamil. On the contrary, EVs did not modify the resistance to cisplatin. Mechanistically, the transfer of P-gp can be partially explained in terms of Rab8 upregulation in the drug-resistant cells upon exposure to vincristine, which leads to increased EV release. Furthermore, the EV uptake in the drug sensitive cells takes place via a clathrin-mediated endocytosis. Importantly, these findings were also confirmed in vivo using a mouse xenograft model [88]. These observations clearly demonstrate the transfer of a fully functional drug transporter via EVs, which also acquires a cellular localization in the target cell compatible with the efflux activity, and strongly suggest increased drug efflux as the mechanism underlying the stimulation of drug resistance by EVs.
Similar conclusions were obtained using osteosarcoma cells (MG-63). Doxorubicin-resistant cells with higher levels of P-gp mRNA produced EVs with higher content of this messenger. Treatment of sensitive MG-63 cells with these EVs resulted in an increase in P-gp mRNA expression in target cells. Concomitantly, an increase in resistance to doxorubicin was observed [89]. Noteworthy, several miRNAs from the drug-resistant cells were also transferred to the sensitive cells via EVs. This way, other cancer-related features such as invasion were modified by EVs. However, to date, there is no evidence relating these miRNAs to P-gp expression [89]. A similar study, also using drug-resistant and drug-sensitive MG-63 cells, reported the transfer of both P-gp and its coding mRNA with the concomitant enhancement of chemoresistance in target cells [90].
EV-mediated transfer of transporters has also been described in cancers of the digestive tract. In the gastric cancer cell lines HGC27 and KATOIII, paclitaxel-resistant clones with P-gp upregulation released EVs with higher P-gp content than sensitive clones. Furthermore, the addition of EVs from the resistant cells to the sensitive counterparts stimulated the resistance to paclitaxel, most likely due to enhanced drug efflux [91]. Also, EVs from drug-resistant HepG2 (i.e., hepatocellular carcinoma) cells increased the resistance of parental HepG2 cells to cisplatin. Mechanistically, EVs ameliorate the increase in ROS triggered by cisplatin. Furthermore, exposure to EVs increased the expression of P-gp in target cells. Similar findings were observed when EVs from resistant HepG2 cells were added to sensitive Huh7 and SMMC-7721 cells, also derived from hepatocellular carcinoma. Since the EVs from the resistant cells exhibited higher P-gp content than EVs from sensitive cells, an EV-mediated transfer of the transporter protein seems feasible [92]. However, whether higher P-gp expression after exposure to EVs is directly related to the enhanced resistance to cisplatin, or whether it belongs to a package of malignancy-related features transferred via EVs, as mentioned above [89], without directly contributing to the resistance to this particular drug has not been demonstrated. Similarly, proteomics analysis identified P-gp enrichment in EVs obtained from the pancreatic juice of patients with pancreatic adenocarcinoma as well as in EVs from pancreatic cancer cell lines capan-1 and MIA PaCa-2. The association between the expression levels of P-gp and therapy resistance was, however, not investigated [93]. Considering that EVs obtained from pancreatic juice are likely to derive from pancreatic tissue, this approach could be used to obtain tissue- and, eventually, tumor-specific information without interference from EVs of other tissues, as is the case for plasma-derived EVs. On the contrary, the sample collection would constitute a highly invasive procedure, the application of which may difficult in clinical practice.
2.2. EV-Mediated Regulation of ABC Transporters
2.2.1. MicroRNAs
Inhibition of Multidrug Resistance via EV-Carried MicroRNAs
Contrary to previous studies, where, mostly, the stimulation of drug-resistance by EVs was described, the opposite effect may also occur upon delivery of EV cargo. This is the case when the vesicles transport miRNAs targeting drug transporters, as demonstrated, for instance, in human acute lymphoblastic leukemia cells (CEM). The P-gp-overexpressing CEM clone (VLB100) releases EVs enriched in miR-326, which, upon addition to the MRP1-overexpressing clone E1000, led to MRP1 downregulation at the mRNA and protein levels. The effect was prevented by transfection of target cells with an miR-326 inhibitor, thus confirming the regulation of MRP1 by this miRNA [94].
Alternatively, miRNAs may regulate drug transporters in a more indirect way, for example, by targeting mRNAs codifying regulatory proteins. This was the case in gastric cancer cells. To establish this mechanism, SGC-7901 and MGC-803 drug-sensitive gastric cancer cells and SGC-7901/5FU cells, resistant to 5-fluorouracil (5-FU) and cisplatin, were used.
Treatment of SGC-791/5FU cells with SGC-7901 and MGC-803 EVs increased sensitivity to 5-FU and cisplatin. The prevention of the effect by treatment of donor cells with GW4869 clearly confirms the involvement of EVs in the transfer of the drug-sensitive phenotype. EVs from both drug-sensitive cells carried significantly higher levels of miR-107 than EVs from the drug-resistant clone. Moreover, miR-107 mimics reproduced the increase in chemosensitivity achieved by treatment of SGC-7901/5FU cells with EVs, further suggesting the sensitizing role of this miRNA. Treatment with SGC-7901 EVs also resulted in the downregulation of P-gp protein expression in target cells. Mechanistically, the authors proposed the downregulation of HMGA2 (high mobility group A2) by binding of miR-107 to its 3′UTR. This results in the inhibition of p-mTOR, which may be responsible for the downregulation of P-gp and increased drug sensitivity [95].
In MG-63 osteosarcoma cells, EVs from parental cells exposed to the flavonoid luteolin enhanced the sensitivity of the resistant clone MG-63/DOX to doxorubicin. Upregulation of miR-384 by luteolin has been proposed as one of the main factors leading to this outcome. In fact, miR-384 targets pleiotrophin (PTN), which may regulate P-gp expression via the β-catenin pathway. Using a mice xenograft model, increase in miR-384, downregulation of P-gp in the tumor cells, and reduced tumor growth due to the treatment with luteolin were also observed [96]. A similar process was proposed in MCF7 cells. Here, EVs from doxorubicin- and docetaxel-resistant clones (MCF7/ADR and MCF7/DOC, respectively) triggered drug resistance in drug-sensitive MCF7 cells. In this experimental setting, β-elemene, a vegetal compound used in traditional Chinese medicine, counteracted the development of resistance. The authors proposed a mechanism consisting in miR-34a upregulation and miR-452 downregulation in the EVs and downregulation of P-gp expression in drug-resistant cells. However, a complete molecular mechanism linking all these observations is still missing [97].
Stimulation of Multidrug Resistance via EV-Carried MicroRNAs
Delivery of miRNAs by EVs may ultimately result in transporter upregulation and development of a multidrug-resistant phenotype. In this regard, a study on breast cancer cell lines proposed miR-423-5p delivered by EVs as an indirect regulator of chemoresistance. In particular, EVs from cisplatin-resistant MDA-MB-231 cells (231/DDP) were added to the culture medium of drug-sensitive MDA-MB-231, MCF7, and SKBR3 breast cancer cells. As a result, an increase in the resistance to cisplatin concomitant with P-gp upregulation was observed. A miRNA analysis pointed to a significant enrichment in miR-423-5p in EVs from 231/DDP cells respect to EVs from the parental cells. In line with this, transfection of drug-sensitive cells with miR-423-5p mimics potentiated the effect of 231/DDP EVs in terms of P-gp upregulation and resistance to cisplatin while, on the contrary, transfection with a miR-423-5p inhibitor counteracted this effect. Altogether, the study clearly shows the stimulation of drug resistance by EVs. While the experimental evidence points to the role of P-gp and its regulation by miR-423-5p in EV-mediated resistance to cisplatin, further studies such as overexpression or silencing of miR-423-5p in donor 231/DDP cells would nicely provide final confirmation about this mechanism [98].
Participation of EV-delivered miR-9-5p, miR-195-5p, and miR-203a-3p in breast cancer multidrug resistance was also demonstrated. Treatment of MDA-MB-231 cells with docetaxel or doxorubicin led to an enrichment in these three miRNAs in EVs. Treatment of parental cells with these EVs induced sphere formation and led to significant downregulation of the transcription factor ONECUT2 (one cut homeobox 2) and the up-regulation of stemness related proteins (i.e., NOTCH1, SOX2, SOX9, NANOG, OCT4). The effects were prevented by simultaneous transfection of target cells with inhibitors of the abovementioned miRNAs. Knockdown experiments demonstrated the upregulation of P-gp, MRP1, and BCRP by decreased levels of ONECUT2. Also, a xenograft model in mice with wild type and Rab27A knockdown MDA-MB-231 cells (i.e., with reduced EV release) showed improved response to docetaxel in mice with deficient EV production. Altogether, the experimental evidence suggests the downregulation of ONECUT2 by miRNAs carried by EVs, which ultimately results in the induction of stemness-related proteins and drug transporters in target cells [99].
Prostate cancer cells also exhibited indirect transporter regulation by miRNAs. In this case, however, the resistance mechanism does not involve the shuttling of EV cargo between cells. Instead, treatment of DU145 cells with fludarabine resulted in changes in the release of different miRNAs to the extracellular space via EVs. While most of the analyzed miRNAs were enriched in EVs from treated cells, a decrease in the EV levels of miR-485-3p was observed. Concomitantly, an increase in the intracellular levels of miR-485-3p was also registered, suggesting a mechanism aimed at the conservation of this specific miRNA in the treated cells. In terms of drug resistance, miR-485-3p accumulation was associated with the downregulation of its target, the β subunit of the nuclear factor Y (NF-YB), which constitutes a negative regulator of P-gp transcription. In this way, a selective decrease in miR-485-3p in EVs and the consequent increased miR-485-3p levels in the cells lead to increased P-gp mRNA expression [100].
In ovarian cancer, EV-mediated transfer of miR-429 from drug-resistant SKOV3 cells to drug-sensitive A2780 led to acquisition of cisplatin resistance in target cells. The mechanism was confirmed using the EV release inhibitor GW4869 as well as a miRNA inhibitor. However, the participation of drug transporting proteins in the resistance mechanism was not investigated [101]. Similarly, EVs from drug-resistant hepatocellular carcinoma Bel7402 stimulated resistance to 5-FU, oxaliplatin, sorafenib, and gemcitabine in parental drug-sensitive Bel7402 cells. This effect was attributed to an enrichment in miR-32-5p in the EVs. Here, the participation of drug transporters also remains uninvestigated [102].
As mentioned in Inhibition of Multidrug Resistance via EV-Carried microRNAs Section, EVs from nontumoral cells may deliver miRNAs to cancer cells. Cases of increased drug resistance in target cells have hereby also been described. Higher levels of miR-301b-3p have been associated with gastric cancer drug resistance in vivo and in vitro. Treatment of SGC7901 gastric cancer cells resistant to cisplatin and vincristine with EVs from mesenchymal stem cells (MSCs) resulted in miR-301b-3p and P-gp upregulation. All the effects were counteracted by transfection of the donor MSC cells with anti-miR-301b-3p. Changes in the expression of drug transporters was attributed to the downregulation of TXNIP (thioredoxin-interacting protein) by EVs [103]. A similar effect, although with other molecular players, was observed in multiple myeloma cells. Here, MSC EVs delivered miR-155 to MPC-11 myeloma cells and increased P-gp, MRP1, and BCRP expression and resistance to bortezomib and dexamethasone. The molecular pathway linking the delivery of miR-155 to target cells and transporter regulation is not fully elucidated [104].
Also, cells from the tumor microenvironment release EVs, which may transfer miRNAs to tumor cells. This was the case in a model of coculture of epithelial ovarian cancer cells with M2 tumor-associated macrophages (M2 TAM). First, coculture of both cell types in a transwell system increased resistance to cisplatin in A2780 and A2780/DDP (i.e., already cisplatin-resistant) cells, suggesting the presence of a diffusible factor mediating cell–cell communication and being responsible for the effect. Treatment of A2780 and A2780/DDP cells with EVs isolated from M2 TAM led to diverse effects in terms of mRNA expression of drug transporters in target cells. Namely, P-gp was upregulated in A2780/DDP cells by treatment with EVs. P-gp upregulation by EVs was also observed in A2780 cells, although only in the presence of cisplatin. MRP1 and BCRP were slightly upregulated by M2 EVs in A2780 cells, while no changes were observed in A2780/DDP cells. miR-221-3p, which was enriched in M2 EVs with respect to monocytes from peripheral blood, was proposed as an important regulator of the drug resistance in this model [105].
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Duan, C.; Yu, M.; Xu, J.; Li, B.Y.; Zhao, Y.; Kankala, R.K. Overcoming Cancer Multi-Drug Resistance (MDR): Reasons, Mechanisms, Nanotherapeutic Solutions, and Challenges. Biomed. Pharmacother. 2023, 162, 114643.
- Hu, T.; Gong, H.; Xu, J.; Huang, Y.; Wu, F.; He, Z. Nanomedicines for Overcoming Cancer Drug Resistance. Pharmaceutics 2022, 14, 1606.
- Ceballos, M.P.; Rigalli, J.P.; Cere, L.I.; Semeniuk, M.; Catania, V.A.; Ruiz, M.L. ABC Transporters: Regulation and Association with Multidrug Resistance in Hepatocellular Carcinoma and Colorectal Carcinoma. Curr. Med. Chem. 2019, 26, 1224–1250.
- Zhang, L.; Ye, B.; Chen, Z.; Chen, Z.S. Progress in the Studies on the Molecular Mechanisms Associated with Multidrug Resistance in Cancers. Acta Pharm. Sin. B 2023, 13, 982–997.
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The Multi-Factorial Nature of Clinical Multidrug Resistance in Cancer. Drug Resist. Updates 2019, 46, 100645.
- Yang, L.; Li, A.; Wang, Y.; Zhang, Y. Intratumoral Microbiota: Roles in Cancer Initiation, Development and Therapeutic Efficacy. Signal Transduct. Target. Ther. 2023, 8, 35.
- Alonso-Peña, M.; Sanchez-Martin, A.; Sanchon-Sanchez, P.; Soto-Muñiz, M.; Espinosa-Escudero, R.; Marin, J.J.G. Pharmacogenetics of Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Drug Resist 2019, 2, 680–709.
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of Tumor Resistance to Immune Checkpoint Blockade and Combination Strategies to Overcome Resistance. Front. Immunol. 2022, 13, 915094.
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the Drug-Microbiota Interactions in Anticancer Therapies. Microbiome 2018, 6, 92.
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local Bacteria Affect the Efficacy of Chemotherapeutic Drugs. Sci. Rep. 2015, 5, 14554.
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744.
- Zhu, G.; Su, H.; Johnson, C.H.; Khan, S.A.; Kluger, H.; Lu, L. Intratumour Microbiome Associated with the Infiltration of Cytotoxic CD8+ T Cells and Patient Survival in Cutaneous Melanoma. Eur. J. Cancer 2021, 151, 25–34.
- Rice, A.J.; Park, A.; Pinkett, H.W. Diversity in ABC Transporters: Type I, II and III Importers. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 426–437.
- Eckenstaler, R.; Benndorf, R.A. 3D Structure of the Transporter ABCG2—What’s New? Br. J. Pharmacol. 2020, 177, 1485–1496.
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC Transporters: The Power to Change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227.
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian Drug Efflux Transporters of the ATP Binding Cassette (ABC) Family in Multidrug Resistance: A Review of the Past Decade. Cancer Lett. 2016, 370, 153–164.
- Beek, J.; Guskov, A.; Slotboom, D.J. Structural Diversity of ABC Transporters. J. Gen. Physiol. 2014, 143, 419–435.
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. BBA Biomembr. 1976, 455, 152–162.
- Jones, P.M.; George, A.M. The ABC Transporter Structure and Mechanism: Perspectives on Recent Research. Cell. Mol. Life Sci. 2004, 61, 682–699.
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41.
- Lu, L.; Katsaros, D.; Wiley, A.; Rigault De La Longrais, I.A.; Puopolo, M.; Yu, H. Expression of MDR1 in Epithelial Ovarian Cancer and Its Association with Disease Progression. Oncol. Res. 2007, 16, 395–403.
- Penson, R.T.; Oliva, E.; Skates, S.J.; Glyptis, T.; Fuller, A.F.; Goodman, A.; Seiden, M.V. Expression of Multidrug Resistance-1 Protein Inversely Correlates with Paclitaxel Response and Survival in Ovarian Cancer Patients: A Study in Serial Samples. Gynecol. Oncol. 2004, 93, 98–106.
- Assaraf, Y.G.; Borgnia, M.J. Differential Reversal of Lipophilic Antifolate Resistance in Mammalian Cells with Modulators of the Multidrug Resistance Phenotype. Anticancer Drugs 1993, 4, 395–406.
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of Atp-Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58.
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236.
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a Novel Raf Kinase Inhibitor. Endocr. Relat. Cancer 2001, 8, 219–225.
- Sauna, Z.E.; Smith, M.M.; Müller, M.; Kerr, K.M.; Ambudkar, S.V. The Mechanism of Action of Multidrug-Resistance-Linked P-Glycoprotein. J. Bioenerg. Biomembr. 2001, 33, 481–491.
- Klimecki, W.T.; Futscher, B.W.; Grogan, T.M.; Dalton, W.S. P-Glycoprotein Expression and Function in Circulating Blood Cells from Normal Volunteers. Blood 1994, 83, 2451–2458.
- Alvarez, M.; Paull, K.; Monks, A.; Hose, C.; Lee, J.S.; Weinstein, J.; Grever, M.; Bates, S.; Fojo, T. Generation of a Drug Resistance Profile by Quantitation of Mdr-1/P-Glycoprotein in the Cell Lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Investig. 1995, 95, 2205–2214.
- Choudhuri, S.; Klaassen, C.D. Structure, Function, Expression, Genomic Organization, and Single Nucleotide Polymorphisms of Human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) Efflux Transporters. Int. J. Toxicol. 2006, 25, 231–259.
- Baker, E.K.; Johnstone, R.W.; Zalcberg, J.R.; El-Osta, A. Epigenetic Changes to the MDR1 Locus in Response to Chemotherapeutic Drugs. Oncogene 2005, 24, 8061–8075.
- Scotto, K.W. Transcriptional Regulation of ABC Drug Transporters. Oncogene 2003, 22, 7496–7511.
- Yakusheva, E.N.; Titov, D.S. Structure and Function of Multidrug Resistance Protein 1. Biochemistry 2018, 83, 907–929.
- Burk, O.; Arnold, K.A.; Geick, A.; Tegude, H.; Eichelbaum, M. A Role for Constitutive Androstane Receptor in the Regulation of Human Intestinal MDR1 Expression. Biol. Chem. 2005, 386, 503–513.
- Rigalli, J.P.; Ciriaci, N.; Arias, A.; Ceballos, M.P.; Villanueva, S.S.M.; Luquita, M.G.; Mottino, A.D.; Ghanem, C.I.I.; Catania, V.A.; Ruiz, M.L. Regulation of Multidrug Resistance Proteins by Genistein in a Hepatocarcinoma Cell Line: Impact on Sorafenib Cytotoxicity. PLoS ONE 2015, 10, e0119502.
- Hu, Y.F.; Qiu, W.; Liu, Z.Q.; Zhu, L.J.; Liu, Z.Q.; Tu, J.H.; Wang, D.; Li, Z.; He, J.; Zhong, G.P.; et al. Effects of Genetic Polymorphisms of CYP3A4, CYP3A5 and MDR1 on Cyclosporine Pharmacokinetics after Renal Transplantation. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1093–1098.
- Jing, W.; Safarpour, Y.; Zhang, T.; Guo, P.; Chen, G.; Wu, X.; Fu, Q.; Wang, Y. Berberine Upregulates P-Glycoprotein in Human Caco-2 Cells and in an Experimental Model of Colitis in the Rat via Activation of Nrf2-Dependent Mechanismss. J. Pharmacol. Exp. Ther. 2018, 366, 332–340.
- Brayboy, L.M.; Knapik, L.O.; Long, S.; Westrick, M.; Wessel, G.M. Ovarian Hormones Modulate Multidrug Resistance Transporters in the Ovary. Contracept. Reprod. Med. 2018, 3, 26.
- Mutoh, K.; Tsukahara, S.; Mitsuhashi, J.; Katayama, K.; Sugimoto, Y. Estrogen-Mediated Post Transcriptional down-Regulation of P-Glycoprotein in MDR1-Transduced Human Breast Cancer Cells. Cancer Sci. 2006, 97, 1198–1204.
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Regulation Exerted by MiRNAs in the Promoter and UTR Sequences: MDR1/P-Gp Expression as a Particular Case. DNA Cell Biol. 2012, 31, 1358–1364.
- Yague, E.; Armesilla, A.L.; Harrison, G.; Elliott, J.; Sardini, A.; Higgins, C.F.; Raguz, S. P-Glycoprotein (MDR1) Expression in Leukemic Cells Is Regulated at Two Distinct Steps, MRNA Stabilization and Translational Initiation. J. Biol. Chem. 2003, 278, 10344–10352.
- Ménez, C.; Mselli-Lakhal, L.; Foucaud-Vignault, M.; Balaguer, P.; Alvinerie, M.; Lespine, A. Ivermectin Induces P-Glycoprotein Expression and Function through MRNA Stabilization in Murine Hepatocyte Cell Line. Biochem. Pharmacol. 2012, 83, 269–278.
- Kool, M.; De Haa, M.; Scheffer, G.L.; Scheper, R.J.; Van Eijk, M.J.T.; Juijn, J.A. Analysis of Expression of CMOAT (MRP2), MRP3, MRP4, and MRP5, Homologues of the Multidrug Resistance-Associated Protein Gene (MRP1), in Human Cancer Cell Lines. Cancer Res. 1997, 57, 3537–3547.
- Hopper, E.; Belinsky, M.G.; Zeng, H.; Tosolini, A.; Testa, J.R.; Kruh, G.D. Analysis of the Structure and Expression Pattern of MRP7 (ABCC10), a New Member of the MRP Subfamily. Cancer Lett. 2001, 162, 181–191.
- Bera, T.K.; Lee, S.; Salvatore, G.; Lee, B.; Pastan, I. MRP8, A New Member of ABC Transporter Superfamily, Identified by EST Database Mining and Gene Prediction Program, Is Highly Expressed in Breast Cancer. Mol. Med. 2001, 7, 409–416.
- Hirohashi, T.; Suzuki, H.; Sugiyama, Y. Characterization of the Transport Properties of Cloned Rat Multidrug Resistance-Associated Protein 3 (MRP3). J. Biol. Chem. 1999, 274, 15181–15185.
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a Transporter Gene in a Multidrug-Resistant Human Lung Cancer Cell Line. Science 1992, 258, 1650–1654.
- Kunická, T.; Souček, P. Importance of ABCC1 for Cancer Therapy and Prognosis. Drug Metab. Rev. 2014, 46, 325–342.
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A Family of Drug Transporters: The Multidrug Resistance-Associated Proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302.
- Büchler, M.; König, J.; Brom, M.; Kartenbeck, J.; Spring, H.; Horie, T.; Keppler, D. CDNA Cloning of the Hepatocyte Canalicular Isoform of the Multidrug Resistance Protein, CMrp, Reveals a Novel Conjugate Export Pump Deficient in Hyperbilirubinemic Mutant Rats. J. Biol. Chem. 1996, 271, 15091–15098.
- Klaassen, C.D.; Aleksunes, L.M. Xenobiotic, Bile Acid, and Cholesterol Transporters: Function and Regulation. Pharmacol. Rev. 2010, 62, 1–96.
- König, J.; Rost, D.; Cui, Y.; Keppler, D. Characterization of the Human Multidrug Resistance Protein Isoform MRP3 Localized to the Basolateral Hepatocyte Membrane. Hepatology 1999, 29, 1156–1163.
- Zelcer, N.; van de Wetering, K.; Hillebrand, M.; Sarton, E.; Kuil, A.; Wielinga, P.R.; Tephly, T.; Dahan, A.; Beijnen, J.H.; Borst, P. Mice Lacking Multidrug Resistance Protein 3 Show Altered Morphine Pharmacokinetics and Morphine-6-Glucuronide Antinociception. Proc. Natl. Acad. Sci. USA 2005, 102, 7274–7279.
- Kuroda, M.; Kobayashi, Y.; Tanaka, Y.; Itani, T.; Mifuji, R.; Araki, J.; Kaito, M.; Adachi, Y. Increased Hepatic and Renal Expressions of Multidrug Resistance-Associated Protein 3 in Eisai Hyperbilirubinuria Rats. J. Gastroenterol. Hepatol. 2004, 19, 146–153.
- Rost, D.; König, J.; Weiss, G.; Klar, E.; Stremmel, W.; Keppler, D. Expression and Localization of the Multidrug Resistance Proteins MRP2 and MRP3 in Human Gallbladder Epithelia. Gastroenterology 2001, 121, 1203–1208.
- Borst, P.; Zelcer, N.; van de Wetering, K. MRP2 and 3 in Health and Disease. Cancer Lett. 2006, 234, 51–61.
- Zollner, G.; Wagner, M.; Fickert, P.; Silbert, D.; Fuchsbichler, A.; Zatloukal, K.; Denk, H.; Trauner, M. Hepatobiliary Transporter Expression in Human Hepatocellular Carcinoma. Liver Int. 2005, 25, 367–379.
- Benderra, Z.; Faussat, A.M.; Sayada, L.; Perrot, J.-Y.; Tang, R.; Chaoui, D.; Morjani, H.; Marzac, C.; Marie, J.-P.; Legrand, O. MRP3, BCRP, and P-Glycoprotein Activities Are Prognostic Factors in Adult Acute Myeloid Leukemia. Clin. Cancer Res. 2005, 11, 7764–7772.
- Shukla, S.; Ohnuma, S.; V. Ambudkar, S. Improving Cancer Chemotherapy with Modulators of ABC Drug Transporters. Curr. Drug Targets 2011, 12, 621–630.
- Shibayama, Y.; Nakano, K.; Maeda, H.; Taguchi, M.; Ikeda, R.; Sugawara, M.; Iseki, K.; Takeda, Y.; Yamada, K. Multidrug Resistance Protein 2 Implicates Anticancer Drug-Resistance to Sorafenib. Biol. Pharm. Bull. 2011, 34, 433–435.
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T. MRP3 as a Novel Resistance Factor for Sorafenib in Hepatocellular Carcinoma. Oncotarget 2016, 7, 7207–7215.
- Schuetz, J.D.; Connelly, M.C.; Sun, D.; Paibir, S.G.; Flynn, P.M.; Srinivas, R.V.; Kumar, A.; Fridland, A. MRP4: A Previously Unidentified Factor in Resistance to Nucleoside-Based Antiviral Drugs. Nat. Med. 1999, 5, 1048–1051.
- Wijnholds, J.; Mol, C.A.; van Deemter, L.; de Haas, M.; Scheffer, G.L.; Baas, F.; Beijnen, J.H.; Scheper, R.J.; Hatse, S.; De Clercq, E.; et al. Multidrug-Resistance Protein 5 Is a Multispecific Organic Anion Transporter Able to Transport Nucleotide Analogs. Proc. Natl. Acad. Sci. USA 2000, 97, 7476–7481.
- Lee, K. Analysis of the MRP4 Drug Resistance Profile in Transfected NIH3T3 Cells. J. Natl. Cancer Inst. 2000, 92, 1934–1940.
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A Multidrug Resistance Transporter from Human MCF-7 Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670.
- Liu, F.-S. Mechanisms of Chemotherapeutic Drug Resistance in Cancer Therapy—A Quick Review. Taiwan J. Obstet. Gynecol. 2009, 48, 239–244.
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 Expression, Function, and Promoter Methylation in Human Multiple Myeloma. Blood 2006, 108, 3881–3889.
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME-Tox). Curr. Drug Deliv. 2008, 1, 379–393.
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792.
- Kim, B.; Fatayer, H.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Valleley, E.M.A.; Verghese, E.T.; Williams, B.J.; Thorne, J.L.; Hughes, T.A. Neoadjuvant Chemotherapy Induces Expression Levels of Breast Cancer Resistance Protein that Predict Disease-Free Survival in Breast Cancer. PLoS ONE 2013, 8, e62766.
- D’Souza-Schorey, C.; Schorey, J.S. Regulation and Mechanisms of Extracellular Vesicle Biogenesis and Secretion. Essays Biochem. 2018, 62, 125–133.
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Yates, A.G.; Pink, R.C.; Erdbrügger, U.; Siljander, P.R.-M.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In Sickness and in Health: The Functional Role of Extracellular Vesicles in Physiology and Pathology in Vivo. Part I: Health and Normal Physiology. J. Extracell. Vesicles 2022, 11, e12151.
- Yates, A.G.; Pink, R.C.; Erdbrügger, U.; Siljander, P.R.-M.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In Sickness and in Health: The Functional Role of Extracellular Vesicles in Physiology and Pathology in Vivo. Part II: Pathology. J. Extracell. Vesicles 2022, 11, e12190.
- Lee, J.; Lee, S.-A.; Gu, N.-Y.; Jeong, S.Y.; Byeon, J.S.; Jeong, D.-U.; Ouh, I.-O.; Lee, Y.-H.; Hyun, B.-H. Canine Natural Killer Cell-Derived Exosomes Exhibit Antitumor Activity in a Mouse Model of Canine Mammary Tumor. Biomed Res. Int. 2021, 2021, 6690704.
- Sousa, D.; Lima, R.T.; Lopes-Rodrigues, V.; Gonzalez, E.; Royo, F.; Xavier, C.P.R.; Falcón-Pérez, J.M.; Vasconcelos, M.H. Different Ability of Multidrug-Resistant and -Sensitive Counterpart Cells to Release and Capture Extracellular Vesicles. Cells 2021, 10, 2886.
- Lopes-Rodrigues, V.; Di Luca, A.; Sousa, D.; Seca, H.; Meleady, P.; Henry, M.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug Resistant Tumour Cells Shed More Microvesicle-like EVs and Less Exosomes than Their Drug-Sensitive Counterpart Cells. Biochim. Biophys. Acta 2016, 1860, 618–627.
- Berguetti, T.S.; Quintaes, L.S.P.; Hancio Pereira, T.; Robaina, M.; Cruz, A.; Maia, R.C.; de Souza, P. TNF-α Modulates P-Glycoprotein Expression and Contributes to Cellular Proliferation via Extracellular Vesicles. Cells 2019, 8, 500.
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different Modalities of Intercellular Membrane Exchanges Mediate Cell-to-Cell p-Glycoprotein Transfers in MCF-7 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 7374–7387.
- Wang, X.; Xu, C.; Hua, Y.; Sun, L.; Cheng, K.; Jia, Z.; Han, Y.; Dong, J.; Cui, Y.; Yang, Z. Exosomes Play an Important Role in the Process of Psoralen Reverse Multidrug Resistance of Breast Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 186.
- Lv, M.; Zhu, X.; Chen, W.; Zhong, S.; Hu, Q.; Ma, T.; Zhang, J.; Chen, L.; Tang, J.; Zhao, J. Exosomes Mediate Drug Resistance Transfer in MCF-7 Breast Cancer Cells and a Probable Mechanism Is Delivery of P-Glycoprotein. Tumor Biol. 2014, 35, 10773–10779.
- Jaiswal, R.; Gong, J.; Sambasivam, S.; Combes, V.; Mathys, J.-M.; Davey, R.; Grau, G.E.R.; Bebawy, M. Microparticle-associated Nucleic Acids Mediate Trait Dominance in Cancer. FASEB J. 2012, 26, 420–429.
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.R.; Bebawy, M. Breast Cancer-Derived Microparticles Display Tissue Selectivity in the Transfer of Resistance Proteins to Cells. PLoS ONE 2013, 8, e61515.
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.-X. Tumor Heterogeneity Reshapes the Tumor Microenvironment to Influence Drug Resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033.
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E.R. Membrane Microparticles Mediate Transfer of P-Glycoprotein to Drug Sensitive Cancer Cells. Leukemia 2009, 23, 1643–1649.
- Wang, X.; Qiao, D.; Chen, L.; Xu, M.; Chen, S.; Huang, L.; Wang, F.; Chen, Z.; Cai, J.; Fu, L. Chemotherapeutic Drugs Stimulate the Release and Recycling of Extracellular Vesicles to Assist Cancer Cells in Developing an Urgent Chemoresistance. Mol. Cancer 2019, 18, 182.
- Cai, T.; Zhang, C.; Zhan, T. Transfer of Exosomal MicroRNAs Confers Doxorubicin Resistance in Osteosarcoma Cells. Mol. Med. Rep. 2023, 27, 86.
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal Transfer of MDR by Exosomes in Human Osteosarcoma. Int. J. Oncol. 2016, 49, 189–196.
- Schirizzi, A.; Contino, M.; Carrieri, L.; Riganti, C.; De Leonardis, G.; Scavo, M.P.; Perrone, M.G.; Miciaccia, M.; Kopecka, J.; Refolo, M.G.; et al. The Multiple Combination of Paclitaxel, Ramucirumab and Elacridar Reverses the Paclitaxel-Mediated Resistance in Gastric Cancer Cell Lines. Front. Oncol. 2023, 13, 1129832.
- Tang, Z.; He, J.; Zou, J.; Yu, S.; Sun, X.; Qin, L. Cisplatin-Resistant HepG2 Cell-Derived Exosomes Transfer Cisplatin Resistance to Cisplatin-Sensitive Cells in HCC. PeerJ 2021, 9, e11200.
- Osteikoetxea, X.; Benke, M.; Rodriguez, M.; Pálóczi, K.; Sódar, B.W.; Szvicsek, Z.; Szabó-Taylor, K.; Vukman, K.V.; Kittel, Á.; Wiener, Z.; et al. Detection and Proteomic Characterization of Extracellular Vesicles in Human Pancreatic Juice. Biochem. Biophys. Res. Commun. 2018, 499, 37–43.
- Lu, J.F.; Pokharel, D.; Bebawy, M. A Novel Mechanism Governing the Transcriptional Regulation of ABC Transporters in MDR Cancer Cells. Drug Deliv. Transl. Res. 2017, 7, 276–285.
- Jiang, L.; Zhang, Y.; Guo, L.; Liu, C.; Wang, P.; Ren, W. Exosomal MicroRNA-107 Reverses Chemotherapeutic Drug Resistance of Gastric Cancer Cells through HMGA2/MTOR/P-Gp Pathway. BMC Cancer 2021, 21, 1290.
- Qin, T.; Zhu, W.; Kan, X.; Li, L.; Wu, D. Luteolin Attenuates the Chemoresistance of Osteosarcoma through Inhibiting the PTN/β-Catenin/MDR1 Signaling Axis by Upregulating MiR-384. J. Bone Oncol. 2022, 34, 100429.
- Zhang, J.; Zhang, H.; Yao, Y.-F.; Zhong, S.-L.; Zhao, J.H.; Tang, J.H. β-Elemene Reverses Chemoresistance of Breast Cancer Cells by Reducing Resistance Transmission via Exosomes. Cell. Physiol. Biochem. 2015, 36, 2274–2286.
- Wang, B.; Zhang, Y.; Ye, M.; Wu, J.; Ma, L.; Chen, H. Cisplatin-Resistant MDA-MB-231 Cell-Derived Exosomes Increase the Resistance of Recipient Cells in an Exosomal MiR-423-5p-Dependent Manner. Curr. Drug Metab. 2019, 20, 804–814.
- Shen, M.; Dong, C.; Ruan, X.; Yan, W.; Cao, M.; Pizzo, D.; Wu, X.; Yang, L.; Liu, L.; Ren, X.; et al. Chemotherapy-Induced Extracellular Vesicle MiRNAs Promote Breast Cancer Stemness by Targeting ONECUT2. Cancer Res. 2019, 79, 3608–3621.
- Lucotti, S.; Rainaldi, G.; Evangelista, M.; Rizzo, M. Fludarabine Treatment Favors the Retention of MiR-485-3p by Prostate Cancer Cells: Implications for Survival. Mol. Cancer 2013, 12, 52.
- Li, T.; Lin, L.; Liu, Q.; Gao, W.; Chen, L.; Sha, C.; Chen, Q.; Xu, W.; Li, Y.; Zhu, X. Exosomal Transfer of MiR-429 Confers Chemoresistance in Epithelial Ovarian Cancer. Am. J. Cancer Res. 2021, 11, 2124–2141.
- Fu, X.; Liu, M.; Qu, S.; Ma, J.; Zhang, Y.; Shi, T.; Wen, H.; Yang, Y.; Wang, S.; Wang, J.; et al. Exosomal MicroRNA-32-5p Induces Multidrug Resistance in Hepatocellular Carcinoma via the PI3K/Akt Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 52.
- Zhu, T.; Hu, Z.; Wang, Z.; Ding, H.; Li, R.; Wang, J.; Wang, G. MicroRNA-301b-3p from Mesenchymal Stem Cells-Derived Extracellular Vesicles Inhibits TXNIP to Promote Multidrug Resistance of Gastric Cancer Cells. Cell Biol. Toxicol. 2022.
- Gao, X.; Zhou, J.; Wang, J.; Dong, X.; Chang, Y.; Jin, Y. Mechanism of Exosomal MiR-155 Derived from Bone Marrow Mesenchymal Stem Cells on Stemness Maintenance and Drug Resistance in Myeloma Cells. J. Orthop. Surg. Res. 2021, 16, 637.
- Zhang, X.; Wang, J.; Liu, N.; Wu, W.; Li, H.; Chen, J.; Guo, X. Molecular Mechanism of CD163(+) Tumor-Associated Macrophage (TAM)-Derived Exosome-Induced Cisplatin Resistance in Ovarian Cancer Ascites. Ann. Transl. Med. 2022, 10, 1014.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
676
Revisions:
2 times
(View History)
Update Date:
02 Aug 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No