Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander Lyubitelev | -- | 12199 | 2023-08-01 15:17:22 | | | |
| 2 | Camila Xu | Meta information modification | 12199 | 2023-08-02 02:20:14 | | | | |
| 3 | Camila Xu | Meta information modification | 12199 | 2023-08-02 02:20:45 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lyubitelev, A.; Studitsky, V. Inhibition of Cancer Development by Natural Plant Polyphenols. Encyclopedia. Available online: https://encyclopedia.pub/entry/47496 (accessed on 27 July 2026).
Lyubitelev A, Studitsky V. Inhibition of Cancer Development by Natural Plant Polyphenols. Encyclopedia. Available at: https://encyclopedia.pub/entry/47496. Accessed July 27, 2026.
Lyubitelev, Alexander, Vasily Studitsky. "Inhibition of Cancer Development by Natural Plant Polyphenols" Encyclopedia, https://encyclopedia.pub/entry/47496 (accessed July 27, 2026).
Lyubitelev, A., & Studitsky, V. (2023, August 01). Inhibition of Cancer Development by Natural Plant Polyphenols. In Encyclopedia. https://encyclopedia.pub/entry/47496
Lyubitelev, Alexander and Vasily Studitsky. "Inhibition of Cancer Development by Natural Plant Polyphenols." Encyclopedia. Web. 01 August, 2023.
Copy Citation
Plant polyphenols are one of the largest groups of secondary metabolites—it includes around 8000 compounds with different structures. Polyphenols may vary significantly by their chemical structure and molecular weight, but they always include at least one aromatic ring and hydroxyl group.
polyphenols
cancer
chemoprevention
mutation
antioxidant
1. Introduction
Malignant tumors are among the longest-known diseases in the mankind history [1]. The most significant progress in cancer research was made during the last century and a half, and today, most of the cancers are considered as a set of acquired genetic disorders that lead to uncontrolled proliferation of the affected cells [2][3]. Despite the significant progress that was achieved in this field, malignant tumors continue to be one of the most important sources of morbidity and mortality throughout the world [4]. One of the most important causes of this situation is significant inter- and intra-tumor genetic heterogeneity. This heterogeneity arises from genetic differences between individuals and from different trajectories of acquiring malignant properties. This variety was described as the hallmarks of cancer; the list of these hallmark properties initially consisted of 6 positions5. Later decades confirmed the applicability of this concept [5][6], and the number of the hallmark properties and cancer-enabling characteristics grew up to 125. Cancer-enabling characteristics, such as genetic and epigenetic instability, chronic inflammation, and altered microbiota, do not comprise malignant phenotype per se, but they may accompany the entire process of tumorigenesis, facilitating malignant transformation.
Acquiring all of these properties requires many large-scale changes in cellular physiology and genetics, which, with the exception of rare cases of chromotrypsis [7], cannot be made as a result of a single mutational event. Instead, malignant transformation happens as a result of a gradual accumulation of mutations during several years, or even decades [8]. This process includes three stages, called initiation, promotion, and progression [9]; each stage corresponds to a different type of lesion formed by affected cells: microscopic tumor at the stage of initiation, benign tumor with well-defined boundaries at the stage of promotion, and an invasive malignant tumor at the stage of progression [10]. At the stage of initiation, the first genetic alteration is acquired by the affected cells, providing them with an initial proliferative advantage. The stage of promotion is characterized by preneoplastic cell proliferation and by accumulation of additional genetic alterations. The stage of progression concludes the formation of a malignant tumor; it is characterized by constant and aggressive cell proliferation, invasiveness, and the ability to metastasize. The exact trajectory of acquisition of all the hallmark properties may not be identical for all cells within the neoplastic lesion, resulting in an intra-tumor genetic heterogeneity. Formation of metastases also contributes to the genetic heterogeneity of malignant tumors in a single organism, as metastases usually form at the distant sites from the primary tumors, where they are exposed to new factors that could be absent in their initial microenvironment [11]. This sequence of events only approximately describes the course of malignant transformation. Thus, in some cases, the metastases start to form long before the malignization process is concluded [12]. A single mutation is not always responsible for the emergence of a single hallmark property: it was established that as few as three mutations were sufficient for malignant transformation, at least for certain tumors [9][13]. Currently, it is considered that the number of tumor-promoting mutations in tumor cells at the early stages of carcinogenesis is relatively small; but as the disease progresses, the number grows accordingly. As the progressive deregulation encompasses more genes and processes in the cell, affected signaling pathways that are involved in supporting the cancer hallmark properties gradually become functionally redundant.
Processes responsible for the development of a malignant tumor are controlled by a small set of mutations for only a limited period of time. During the rest of the malignant transformation, its progression is driven by a broad spectrum of factors that simultaneously affect a multitude of cellular components and processes. At the same time, a number of anticancer drugs that are currently used and developed are characterized by a high specificity to their targets, and, although these drugs are able to slow tumor growth or even cause a significant reduction in its volume, their efficiency decreases drastically during long-term use. Such a decrease is caused by intra-tumor genetic heterogeneity, as the small number of malignant cells that are insensitive to the selected drug already exist within the tumor at the beginning of the therapy. Even a combination of several highly specific anticancer drugs cannot always prevent the emergence of this effect. Development of anticancer drugs with multiple molecular targets was proposed in order to overcome this drawback [14][15]. Another group of anticancer drugs that are currently in use in clinical practice consists of substances capable of damaging cellular DNA or inhibiting cell division. While their activity is not limited to a certain molecular target, they lack the selectivity for cancer cells, and the alterations of the genome of normal cells caused by these substances often lead to the formation of therapy-induced tumors.
One approach to reduce the cancer-caused mortality is to prevent the development of a malignancy rather than trying to cure it. It is established that around 45% of cancer cases are preventable [16]. The most straightforward approach to cancer prevention is avoidance of exposure to the environmental carcinogenic factors. This approach, in different forms, is applied in many countries around the world. However, not every environmental factor that is potentially carcinogenic, can be effectively avoided, either because of its ubiquitous nature, or because this particular factor is not yet identified as carcinogenic. Moreover, only a limited fraction of mutations that drive the process of carcinogenesis can be attributed to the environmental factors, with an average of around 29% of driver mutations that are caused by the external sources [17].
Cell-intrinsic processes might also be the cause of cellular DNA damage. Aside from mistakes made by replicative DNA polymerase during each S-period, there are also a multitude of sources of highly reactive compounds within the cells, such as reactive oxygen species [18]. To effectively mitigate DNA-damaging activity of both intrinsic and environmental factors, preventive measures should last for a long period of time, as malignant transformation and formation of metastases take years or even decades to conclude. The concept of cancer chemoprevention implies that such mitigation can be achieved either by neutralizing the DNA-damaging factors with chemopreventive drugs, thus abrogating initiation of tumorigenesis, or by preventing accumulation of the changes in cellular physiology, resulting in deceleration or even halting the promotion and progression stages of malignant transformation [19]. Chemopreventive compounds that abrogate the initiation step of malignant transformation are referred to as blockers. Agents that impede the transition of a pre-neoplastic lesion through the promotion and progression stages are known as suppressors. Chemopreventive compounds differ not only by their mechanisms of action, but also by the stage of the malignant transformation at which they act. Based on this criterion, these agents are classified into primary, secondary, and tertiary [20]. Primary chemopreventors suppress the formation of tumors, secondary chemopreventors are aimed at suppressing the transition of a benign pre-neoplastic lesion into malignant one, while tertiary chemopreventors reduce the risk of tumor recurrence after the successful therapeutic intervention. Thus, a potential chemopreventive drug should be suited for a long-term application, be well tolerated by the human organism, and should have a high toxic-to-therapeutic dose ratio.
When discussing anticancer chemotherapeutic and chemopreventive drugs, their cost is also a concern. Indeed, the annual cost of a chemotherapeutic anticancer drug may be as high as USD 100,000 annually [21]. For chemopreventive drugs, which are typically used for a long period of time, the problem of cost efficiency is of even higher relevance.
2. Structure and Properties of Plant Polyphenols
One of the most prominent features of plant biochemistry, which is almost completely absent in the animal kingdom, is the ability of plants to synthesize a broad spectrum of small organic molecules of multiple different structures, which are collectively known as plant secondary metabolites [22]. More than 200,000 different members of this group are currently known [23]. Secondary metabolites carry out a variety of functions, which include protection from ultraviolet radiation, regulation of physiological processes, resistance to physical stress, and interaction with different organisms, such as herbivorous animals or parasitic fungi [24]. Plant polyphenols are one of the largest groups of secondary metabolites—it includes around 8000 compounds with different structures [25]. Polyphenols may vary significantly by their chemical structure and molecular weight, but they always include at least one aromatic ring and hydroxyl group [26]. Several different variants of classification of the phenolic compounds were proposed. The most widely accepted of these variants divides plant phenolic compounds into two groups: flavonoids and non-flavonoids [27] (Figure 1). Flavonoids are derived from aromatic amino acids, and share a common tricyclic C6-C3-C6 structure [28]. Flavonoids are further divided into flavonols, flavones, isoflavones, flavanones, flavanols, anthocyanins, and anthocyanidins [29]. The group of non-flavonoid polyphenols includes phenolic acids, xanthones, stilbenes, tannins, and lignans [30] (Figure 1).
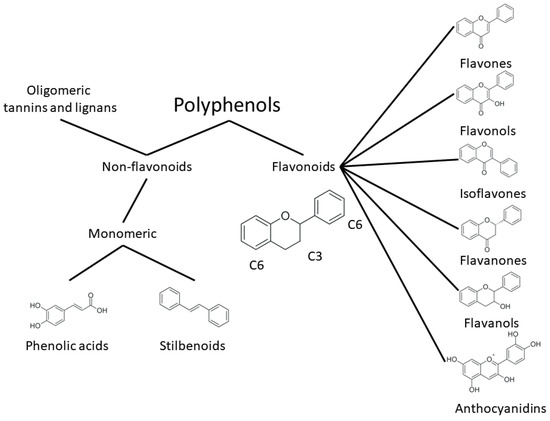
Figure 1. Structure and classification of plant phenolic compounds.
Polyphenolic compounds can be found in large quantities in fruits, seeds, and other edible parts of plants [31][32]. Fruits, berries, legumes, and other plant-derived foods and beverages [33], such as tea [34], wine [35], olive oil [36], or seasonings [37], are rich with polyphenols. Medicinal plants are another source of polyphenolic compounds [38]. The amount of polyphenols in a diet may differ drastically for different regions of the planet [39][40]. Epidemiological studies of the influence of a polyphenol-rich diet on human health suggest that the regular consumption of these compounds is associated with a lower incidence of a multitude of serious chronic diseases, such as metabolic syndrome [41], type II diabetes mellitus [42], neurodegenerative amyloidosis [43], cardiovascular diseases [44], and cancer [45].
As malignant transformation includes several stages and requires a long period of time to be completed [8], decrease in the incidence of cancer observed for individuals on a polyphenol-rich diet may be caused by the anticancer or chemopreventive properties of these compounds. Further experiments with tumor cell cultures and models, as well as clinical studies, established that polyphenolic compounds possess both of these activities [46][47]. For example, the treatment of different tumor cell cultures with plant phenolic compounds revealed the antiproliferative, antiangiogenic, cell cycle-arresting, and proapoptotic activity of these substances [48]. In particular, proapoptotic and antiproliferative activity of polyphenols was demonstrated for the MCF-7 breast cancer cell line [49][50], as well as for several prostate cancer cell lines [51]. Antiangiogenic activity of plant phenolics was demonstrated for murine [52] and human [53] healthy and tumor cell lines treated with these compounds. Anticancer and chemopreventive activity of polyphenols was confirmed by the results of in vivo experiments involving animal-induced tumorigenesis and tumor xenografts [54][55]. Treatment with polyphenols lowered the incidence of neoplastic lesions after initiation of tumorigenesis by N-nitrosobis(2-oxopropyl)amine [56] and suppressed the development of tumors stimulated by 1,2-dimethylhydrazine [57] and azoxymethane [58]. A similar treatment of the animals carrying tumor xenografts led to a significant reduction in tumor volumes and tumor growth rates [59][60]. Finally, the effectiveness of plant phenolic compounds as potential anticancer and chemopreventive drugs was confirmed by several clinical studies [61][62]. Phenolic compounds have antitumor activities not only alone [63][64], but also in combinations with other anticancer drugs [65][66]. At the same time, there are clinical studies in which plant polyphenols failed to demonstrate any anticancer or chemopreventive activity [67][68]. Some studies suggest that polyphenols might even facilitate tumorigenesis [69].
Confirmation of the anticancer and chemopreventive properties of polyphenols brought forth a question about the exact molecular mechanisms that underlie these activities. Unlike many other clinically used plant secondary metabolites that can interact with a limited number of molecular targets, or even with only a single one, plant phenolic compounds are able to interact with a broad spectrum of biological macromolecules of several different classes [70]. It was demonstrated that the anticancer activity of polyphenols might be determined by their specific interaction with signaling [71], catalytic [72], regulatory [73], and receptor [74][75] proteins, as well as by their non-specific chaperone activity [76]. The ability of phenolic compounds to specifically accumulate in cellular membrane lipid rafts, thus causing changes in cell surface receptor protein expression, were also demonstrated [77]. Nucleic acids are yet another molecular target of polyphenols. Polyphenols are not only able to bind to dsDNA, acting as intercalators [78], or major and minor groove ligands [79], but they also may interact with nucleic acid having secondary structures, such as tRNAs [80] or guanine quadruplexes [81].
Evidence of the biological activity of plant phenolic compounds accumulated to date, combined with data regarding their interaction with cellular targets, makes it possible to consider these substances as a basis for the development of perspective anticancer and chemopreventive drugs. Nevertheless, mostly non-specific binding of polyphenols to their molecular targets and a broad range of these targets make it necessary to extensively study the influence of polyphenols on regulatory pathways and processes involved in tumorigenesis and the development of cancer-enabling characteristics.
3. Genetic Instability
Human cells are exposed to a wide variety of DNA-damaging substances, both exogenous and endogenous, which include polycyclic aromatic hydrocarbons [82], polycyclic aromatic amines [83], and a number of other genotoxic agents of various chemical natures [84][85]. Some of them are carcinogens per se, while others are procarcinogens, and may exert carcinogenic properties only after they underwent metabolic activation. Metabolism of procarcinogens, as well as the other xenobiotics, consists of three sequential stages: metabolic activation, conjugation, and excretion [86]. Aromatic hydrocarbon receptor AhR plays a key role in the activation of the xenobiotic metabolism by regulating expression of metabolic activation and conjugation enzymes [87][88] (Figure 2). Binding AhR to its ligand causes a conformational change, thus leading to exposure of a nuclear translocation signal, which is covered by molecules of chaperones, such as p23 or Hsp90, while AhR is in an inactive state. The nuclear translocation signal mediates transfer of the AhR-chaperone complex to the nucleus, where chaperones dissociate from AhR, and the receptor, in turn, forms a heterodimer with Arnt. In this heterodimeric state, AhR is able to bind xenobiotic response elements (XREs) on cell DNA, thus activating the expression of enzymes involved in xenobiotic metabolism. This receptor is also involved in regulating inflammatory response, reaction to hypoxia, and cell cycle progression [89]. Organic compounds with several aromatic rings, including plant polyphenols, are the main ligands of AhR. Direct interaction between AhR and polyphenols was demonstrated for many compounds of this group [90]. The binding of polyphenols to AhR can modulate activity of this receptor by several different mechanisms (Figure 2); the most extensively studied among these is direct competitive inhibition. It was demonstrated that a number of polyphenolic compounds, namely curcumin [91], quercitin [92], and resveratrol [93], together with flavone, apigenin, luteolin, and galangin [94], may prevent AhR activation by polycyclic organic ligands in cellular extracts. Effective inhibitory concentrations of polyphenols in these experiments ranged from 0.019 to 50 µM. Kaempferol, luteolin, apigenin, and fisetin were able to prevent dioxin-mediated activation of AhR-regulated CYP1A1 gene in Caco2 human colon carcinoma cell culture. At the same time, treatment of the same cell culture with quercitin, robinetin, morin, and taxifolin alone, or in combination with dioxin, did not cause any reduction in AhR-regulated genes expression. Moreover, expression of these genes was significantly increased, suggesting that these phenolic compounds act as AhR agonists [95]. It was also demonstrated that quercitin, naringin, hesperidin, and hesperitin were able to enhance luciferase gene expression, which was put under AhR transcriptional control in genetically engineered reporter H1L6.1c2 cell culture [96]. Authors noted that only polyphenols with five hydroxyl residues in their structure were able to act as AhR agonists, whereas phenolic compounds with a different number of hydroxyls acted as antagonists or showed no activity towards AhR. Analysis of the interactions between plant phenolics and AhR made possible the development of molecular models that can predict in vitro agonistic activity of certain types of polyphenols towards this receptor protein [97]. Nevertheless, the principles that govern type and intensity of AhR modulation by plant phenolic molecules are yet to be fully understood [98].
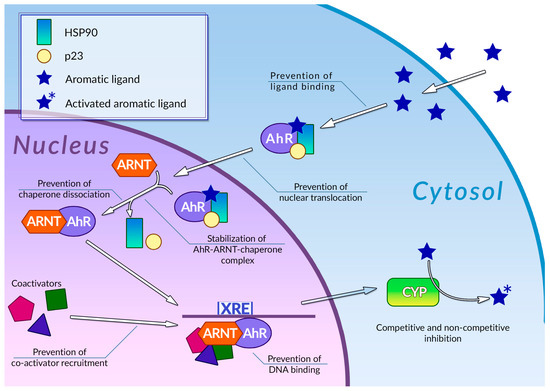
Figure 2. Regulation of phase 1 xenobiotic metabolism and its inhibition by plant polyphenols. See text for detail.
Mechanisms of modulation of AhR activity by polyphenols are not limited to direct competitive inhibition or agonism (Figure 2). Phenolic compounds can also affect a number of stages of the AhR signal transduction pathway. It was demonstrated that galangin, luteolin, apigenin, flavone, kaempferol [99], curcumin [100], and resveratrol [93] in concentrations around 5–50 µM were able to prevent the nuclear translocation of AhR in MCF-7 and Hepa-1c1c7 cell cultures. It was also shown that treatment of Hepa-1c1c7 cells with polyphenols can prevent formation of an active AhR-Arnt heterodimer: this ability was also demonstrated for galangin [101], naringenin, kaempferol, apigenin [99], and epigallocatechin gallate (EGCG) [99][102]. These compounds block AhR-Arnt heterodimer by different mechanisms: EGCG and galangin prevent association of AhR and Arnt by blocking the dissociation of chaperones from AhR. Apigenin and kaempferol stabilize the ternary complex between AhR, Arnt, and chaperones, while naringenin, kaempferol, apigenin, and EGCG were able to prevent Arnt phosphorylation, which is necessary for dimerization. The binding of polyphenols to active AhR-Arnt dimer may also inhibit activation of AhR-regulated genes by preventing binding of the dimer to the XRE DNA elements: the ability of preventing binding of the dimer to the XREs in tumor cell cultures was demonstrated for resveratrol [103], curcumin [100], quercitin, kaempferol [92], and galangin [101]. Plant phenolic compounds may also prevent activation of AhR-responsible genes by inhibiting recruitment of coregulators, such as the ERα estrogen receptor. This ability was demonstrated for resveratrol [93], genistein [104], and kaempferol [105], but the extent of the effect of this inhibition on the efficiency of the activation of transcription is yet to be determined. It is also noteworthy that in some cases, treatment with polyphenols instead of inhibition caused an enhancement of certain stages of the AhR signal transduction pathway: thus, treatment of Hepa-1c1c7 cells with curcumin facilitated nuclear translocation of ligand-bound AhR [100]. At the same time, curcumin efficiently inhibits subsequent stages of AhR activation, such as AhR-Arnt heterodimer formation. The mechanisms of plant polyphenol action on the AhR signal transduction pathway are summarized in Table 1.
Table 1. Mechanisms of action of plant polyphenols on the AhR signal transduction pathway.
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin | Phenolic acid homodimer | Direct competitive inhibition of AhR in cell-free extracts | [91] |
| Quercetin | Flavonol | [92] | |
| Resveratrol | Stilbenoid | [93] | |
| Flavone | Flavone | [94] | |
| Luteolin | Flavone | ||
| Apigenin | Flavone | ||
| Galangin | Flavonol | ||
| Kaempferol | Flavonol | Inhibition of AhR in Caco2 cell culture | [95] |
| Luteolin | Flavone | ||
| Apigenin | Flavone | ||
| Fisetin | Flavonol | ||
| Quercetin | Flavonol | Agonism of AhR in Caco2 cell culture | |
| Robinetin | Flavone | ||
| Taxifolin | Flavonol | ||
| Morin | Flavone | ||
| Quercetin | Flavonol | Stimulation of AhR-regulated luciferase gene expression in H1L6.1c2 reporter cell culture | [96] |
| Naringin | Glycosylated flavanone | ||
| Hesperidin | Glycosylated flavanone | ||
| Hesperitin | Flavanone | ||
| Galangin | Flavonol | Prevention of nuclear translocation of AhR in MCF-7 and Hepa-1c1c7 cell cultures | [99] |
| Apigenin | Flavone | ||
| Luteolin | Flavone | ||
| Flavone | Flavone | ||
| Kaempferol | Flavonol | ||
| Curcumin | Phenolic acid homodimer | [100] | |
| Resveratrol | Stilbenoid | [93] | |
| Naringenin | Flavanone | Prevention of active AhR-Arnt heterodimer formation in cell cultures | [99] |
| Apigenin | Flavone | ||
| Kaempferol | Flavonol | ||
| Galangin | Flavonol | [101] | |
| EGCG | Flavanol and phenolic acid heterodimer | [99][102] | |
| Curcumin | Phenolic acid homodimer | Prevention of XRE binding by AhR-Arnt dimer in tumor cell cultures | [100] |
| Galangin | Flavonol | [101] | |
| Resveratrol | Stilbenoid | [103] | |
| Quercetin | Flavonol | [92] | |
| Kaempferol | Flavonol | ||
| Resveratrol | Stilbenoid | Prevention of AhR-Arnt coactivator recruitment | [93] |
| Genistein | Isoflavone | [104] | |
| Kaempferol | Flavonol | [105] |
The most important proteins, the expression of which is regulated by the AhR signaling pathway, are several proteins of the cytochrome P450 family. The human genome contains more than 50 genes of these redox enzymes. These enzymes participate not only in xenobiotic activation, but also in fatty acid and cholesterol biosynthesis, the metabolism of steroid hormones and vitamins, and unsaturated fatty acid oxidation [106]. Reactions of the metabolic activation of small organic molecules, which are responsible for procarcinogen activation, are catalyzed by cytochromes of subfamilies 1, 2, and 3; CYP1A1, A2 and A6, CYP2A13, CYP2B6, CYP2C9, and CYP3A4 are the most important among them [107]. Plant polyphenols can modulate activity of proteins of this family not only by affecting regulation of their expression, but also by interacting directly with the molecules. Using a combination of spectroscopic methods and molecular modeling approaches it was established that a large number of natural flavonoids is capable of binding to the active site of cytochromesCYP1A1, 1A2, 1B1, 2C9 and 3A4 [108]. For the latter two isoforms it was also found out that inhibition of their activity by flavonoid molecules is carried out by both competitive and non-competitive mechanisms [109]. It is noteworthy that for some flavonoids, such as amentoflavone, apigenin, and galangin, their half-maximal inhibitory concentration measured during this experiment was significantly lower than their previously determined concentration in human plasma, which suggests that the inhibitory activity of these polyphenols is physiologically relevant.
Plant polyphenols are not only able to prevent DNA damage, but also facilitate their detection and repair. For example, treatment with natural phenolic compounds might affect expression of the γH2AX histone variant that marks double-stranded DNA breaks in interphase chromatin, thus leading either to repair of these breaks [110] or to sensitization of tumors to DNA-damaging therapeutic agents [111]. Treatment with these compounds might stimulate DNA repair activity in normal tissues and promote apoptosis in tumor cells [112]. The latter type of activity suggests that natural phenolic compounds may enhance the efficiency of DNA-damaging anticancer therapy. Recent experimental results confirm the higher efficiency of this combined therapy [113].
Organic and inorganic free radicals are other important classes of DNA-damaging substances [114][115]. These compounds are not only capable of damaging DNA molecules by themselves, but they may also react with procarcinogens, thus activating them [116]. The main sources of free radicals in the cell are metabolic reactions and exposure to electromagnetic radiation. They may also form as by-products of the reactions catalyzed by ions of transition metals, such as iron and copper [117]. Free radicals can also damage cellular proteins, yielding them unable to carry out their functions [118]. As formation of the free radicals is unavoidable, living organisms had to develop effective measures to counteract their deleterious effects, such as redox enzymes and their cofactors, which can react with free radicals to neutralize them [119]. Numerous hydroxyl groups and certain other structural features determine the ability of plant polyphenols to react with free radicals, thus effectively scavenging them [120][121]. There are four known types of these reactions: three of them are based on proton and electron transfer from phenolic molecules to free radicals by different mechanisms; the fourth type includes adducts formed after interaction between polyphenol and a free radical [122]. Catechins and their derivatives, as well as plant extracts containing these substances, have the ability to effectively neutralize organic radicals [123] and reactive oxygen and nitrogen species [123][124] in vitro. EGCG has antioxidant activity in vivo, significantly reducing the effect of the induction of inorganic radical formation by sodium fluoride [125]. Gallic acid was able to reduce the severity of dimethylhydrazine-induced oxidative stress in rats, thus preventing induction of colon carcinoma by this carcinogenic compound [126]. Quercitin has a similar chemopreventive activity; it works by effectively mitigating the oxidative stress after induction of prostate carcinoma by high doses of testosterone in Sprague–Dawley rats [127]. Phenolic compounds from an extract of green tea leaves were able to increase the antioxidant potential of human plasma [128][129]. According to numerous clinical studies, consumption of anthocyanins and anthocyanin-rich dietary substances led to a significant decrease in the concentration of oxidative stress biomarkers, such as malonyldialdehyde and oxidized low-density lipoproteins [130]. Increase in the activity of gluthatione peroxidase and superoxide dismutase, as well as a general increase in antioxidant capacity, was also observed. These effects were more prominent for patients with diseases than for healthy volunteers.
Plant polyphenols can mitigate the oxidative stress not only by free radical scavenging, but also by preventing their formation. It was established that phenolic compounds can act as chelators of metal ions, thus decreasing the rate of free radical formation catalyzed by these ions. This kind of activity was demonstrated for flavonoids [131] and for non-flavonoid plant phenolic compounds, including phenolic acids, the structures of which include only a single aromatic ring [132].
Despite the significant amount of evidence of the antioxidant activity of plant polyphenols obtained during the experiments with cell-free extracts, cell cultures, and in vivo studies, the influence of natural phenolic compounds on the cellular redox status turned out to be more complex, and even prooxidant [133]. Polyphenols exerted their prooxidant activity in high concentrations, under high pH values, or in the presence of ions of certain transition metals [134]. Paradoxically, this pro-oxidant activity may also result in an increase in cellular antioxidant defenses: under physiologically relevant concentrations of polyphenols, the amount of reactive oxygen species that are formed by these reactions is not sufficient to induce any significant DNA damage, but enough to elicit cellular mechanisms of protection against the oxidative stress [135]. It is noteworthy that EGCG, previously viewed as a potent antioxidant, demonstrated pro-oxidant activity in this research. It was also established that certain cultivated cancer cells with metabolism and signaling networks distorted by malignant transformation react to the treatment with polyphenols in the opposite way as normal cells. Instead of lowering the severity of oxidative stress and decreasing the concentration of free radicals, this treatment led to a decrease in proliferative activity and to an induction of apoptosis, as the concentration of free radicals was increased to cytotoxic values [134].
Antioxidant activity of plant phenolic compounds is not limited to direct radical scavenging. Polyphenols can also affect the redox status of the cell by attenuating metabolic processes involved in the generation and neutralization of free radicals. Nrf2 is one of the most important transcription factors responsible for activation of the expression of oxidative stress response proteins. Oxidative stress induces dissociation of the Keap1-Nrf2 complex, preventing its Keap1 ubiquitination-induced proteasomal degradation, and thus leading to an accumulation of active Nrf2 and to the activation of the transcription of Nrf2-regulated genes [136]. It was established that quercitin may prevent ubiquitination of Keap1, facilitating accumulation and activation of Nrf2 [137]. EGCG was able to enhance Nrf2 activity in lung tissue in rats, abrogating fluoride-induced oxidative stress [125]. Results of molecular docking suggest that this effect could be determined by the interaction between EGCG and Keap1, preventing this protein from binding to Nrf2 and mediating its proteasome degradation. Similar results were obtained during treatment of L02 human hepatocyte culture with gallic acid, where this phenolic compound mitigated tert-butyl hydroperoxide-induced oxidative stress by preventing formation of the Nrf2-Keap1 complex [138]. Apigenin also has the ability to facilitate accumulation of Nrf2 in the cell, and to prevent high-fructose diet-induced oxidative stress [139]. Molecular modeling of interactions between apigenin and Keap1 suggest that these molecules may form a stable complex that is unable to bind to Nrf2.
Plant polyphenols can also modulate the activity of other proteins involved in the maintenance of the cellular redox status. Green tea polyphenols have an inhibitory activity towards expression of NADPH oxidase subunits, thus attenuating production of reactive oxygen species. The ability of EGCG and theaflavin-3,3′-digallate to inhibit lipopolysaccharide (LPS)-induced induction of inducible nitric oxide synthase (iNOS) expression was also established [140][141].
When considering the antioxidant activity of polyphenols for anticancer and chemopreventive applications, it should be kept in mind that reactive oxygen species are involved in regulation of a number of cellular processes, and in turn, are subjected to strict regulation [142]. There is also evidence that the mode of action of natural antioxidants in vivo differs significantly from the mechanisms identified by in vitro experiments [143]. These concerns raised doubts about the efficiency of high doses of natural antioxidants as anticancer measures [144], especially in combination with the other therapeutic approaches, whose mechanisms of action are based on induction of free radical formation in cancer cells [145][146]. All these complications demand separation of the antitumor and chemopreventive applications of polyphenolic antioxidants [147].
4. Epigenetic Reprogramming
DNA damage imposed by different factors affects the expression and functions of cellular proteins in a straightforward manner—by changing DNA sequence and thus affecting activity of the encoded proteins or misregulating their expression. However, the activity of the regulatory elements of the cellular genome is regulated not only by DNA sequence, but also by a number of non-genomic hereditable markers, commonly known as epigenetic factors. These factors include methylation of DNA, methylation, acetylation, and other post-translational modifications of histones, histone variants incorporated into chromatin, and non-coding RNAs [148]. It was established that disorders of epigenetic regulation may lead to development of one or more cancer hallmark properties, marking epigenetic instability as a cancer-enabling characteristic [149][150]. For example, global DNA hypomethylation found in many cancer cells enhances transcriptional activity in a non-selective manner, whereas hypermethylation of regulatory sequences of tumor suppressor genes may lead to activation of a number of genes and thus facilitate the malignant transformation [151]. Disorders of epigenetic labeling of histone writers, readers, or erasers lead to changes in interphase chromatin compaction and activity and affect gene expression [152]. Chromatin structure and activity is also regulated by the inclusion of different nucleosome core [153][154] and linker [155][156] histone variants. Aberrant activity of microRNA may facilitate expression of pro-oncogenic signaling pathway proteins or inhibit expression of tumor suppressor proteins at a translational level [157]. Finally, guanine quadruplexes might represent yet another system of epigenetic regulation, as suggested by the reverse correlation between the number of folded quadruplexes and the degree of cellular differentiation [158].
Plant phenolic compounds affect a multitude of cellular processes involved in epigenetic regulation. EGCG might attenuate activity of DNMT DNA methyltransferases by downregulating their expression [159], and by inhibiting their catalytic activity [160] after binding directly to these proteins [161][162]. Attenuation of the activity of methyltransferases was detected in cultures of esophageal carcinoma (KYSE 150), colon carcinoma (HT29), prostate adenocarcinoma (PC3), and breast cancer (MCF-7 andMDA-MB-231) cell lines, as well as for tumor xenografts, and led to reactivation of tumor suppressor genes silenced by hypermethylation. Curcumin was also able to rescue the expression of tumor suppressor genes by reversing their hypermethylation. Treatment of cultures of lung (A549 and H460) [163] and breast (MCF-7) [164] cancer cells with this phenolic compound led to a decrease in DNMT3b and DNMT1 methyltransferase expression, respectively. It is noteworthy that the inhibition of hypermethylation was detected at lower concentrations of this polyphenol, starting from 10 µM [165]. Treatment with quercitin potentiated the inhibitory activity of curcumin towards DNA methyltransferases of PC3 and DU145 prostate cancer cells: simultaneous treatment with both compounds inhibited the methyltransferase activity more efficiently than treatment with either substance separately [166]. Other plant polyphenols may also affect the DNA methylation. It was found that resveratrol, applied to MDA-MB-157 breast cancer cell culture in combination with pterostilbene, can decrease the levels of methylation of certain regions of genome by inhibiting the recruiting of DNA methyltransferases by STRT1 histone deacetylase [167]. Resveratrol was also able to selectively reverse hypermethylation of regulatory regions of tumor suppressor genes in MDA-MB-231 breast cancer cell culture [168]. Similar activity towards the RASSF-1α tumor suppressor gene was registered during clinical studies [169]. Remarkably, without pterostilbene, resveratrol has antiproliferative activity towards the MCF10CA1a breast cancer cell culture by the opposite mechanism: treatment with this phenolic compound led to recruitment of DNMT3b methyltransferase for the regulatory sequences of oncogenes, thus attenuating their expression by hypermethylation. Genistein also exerted suppression of DNA methyltransferase activity towards renal (A498, ACHN) [170], prostate (LNCaP, PC3) [171], and breast (MDA-MB-468, MCF-7) cancer cell lines; for the latter cultures, the inhibitory concentration of this phenolic compound was as low as 3.125 µM [172].
Plant polyphenols can also modulate activity of proteins involved in the epigenetic modification of histones in tumor cells. It was established that treatment of LNCaP [173] prostate and RKO, HCT-116 иHT-29 [174] colon cancer cell cultures with EGCG inhibited expression of HDAC 1-3 histone deacetylases. Treatment of HeLa cell culture with this polyphenol did not inhibit histone deacetylases, but instead resulted in the inhibition of histone acetyltransferase activity [175]. Curcumin-inhibited histone deacetylases, as treatment of Raji lymphoblast and several medulloblastoma cell cultures with this polyphenol, suppressed their proliferation by downregulating expression of histone deacetylases [176][177]. Quercitin also exerted modulatory activity towards both histone acetyltransferases and deacetylases, as treatment of HL-60 human leukemia cell culture led to inhibition of deacetylases and to an increase in acetyltransferase activity [178]. At the same time, treatment of MDA-MB-231 and MCF-7 human breast cancer cell cultures resulted in the inhibition of activity of p300 acetyltransferase [179]. Resveratrol can enhance activity of SIRT1 histone deacetylase by binding to its active site, thus leading to the inhibition of the proliferation of breast cancer cells derived from BRCA-1-knockout mice [180]. This plant phenolic compound can also attenuate one of the multiple activities of NuRD histone deacetylase complex by destabilizing its interaction with MTA1 coregulator protein, thus increasing the concentration of active acetylated forms of p53 and PTEN tumor suppressors [181][182]. Treatment with genistein promotes acetylation of histones at the transcription start sites of p16 and p21 tumor suppressor genes in prostate (LNCaP and DuPro) [183] and breast (MDA-MB-231) [184] cancer cell cultures, and of BTG3 tumor suppressor genes in renal (A498, ACHN) [170] and prostate (LNCaP, PC3) [171] cancer cell cultures.
Epigenetic mechanisms can regulate the expression of cellular genes both at the stage of their transcription and by changing the amount or activity of its mRNA. The key components of these regulatory systems are non-coding RNAs (ncRNA) known as microRNAs (miRNAs), capable of inhibiting translation of mRNAs or promoting their degradation [185]. There is a number of other regulatory ncRNAs, such as long or circular ncRNAs, whose regulatory activities towards gene expression are based on different mechanisms [186]. It is well established that aberrant miRNA activity in the cell might promote malignant transformation, as these molecules are involved in regulation of the expression of numerous oncogenes and tumor suppressors [187]. Clinical relevance of non-coding RNAs increases steadily during the last few years; a number of clinical studies consider these regulatory molecules as biomarkers or therapeutic targets [188].
Many plant polyphenols can modulate expression of ncRNA, thus affecting their regulatory activity. It was established that treatment of HepG2 human hepatocellular carcinoma cell culture with EGCG downregulated the expression of 48 different miRNAs, while upregulating expression of another 13 miRNAs at the same time. Upregulated miRNAs included miR-16, which targets mRNA encoding Bcl-2 antiapoptotic protein, causing suppression of the proliferation and induction of apoptosis [189]. Treatment of SK-N-BE2 and IMR-32 malignant human neuroblastoma cell cultures with this phenolic compound suppressed expression of oncogenic miR-92, miR-93, and miR-106b miRNAs, and upregulated expression of tumor suppressors miR-7-1, miR-34a, and miR-99a, causing suppression of the proliferation and induction of apoptosis; treatment with N-(-4-hydroxyphenil)-retinamide potentiated this effect [190]. In CL13 murine lung adenocarcinoma and H1299 human non-small cell lung carcinoma cell cultures, treatment with EGCG suppressed proliferation by upregulating expression of miR-210 caused by the binding of EGCG to hypoxia-induced transcription factor HIF-1α, resulting in its stabilization [191]. This plant phenolic compound can also induce apoptosis in MCF-7 human breast cancer cell culture and mouse xenografts by suppressing the expression of miR-25 [192]. The tumor suppressor miRNAs of the let-7 family is also affected by EGCG: proliferation of human NCI-H446 small-cell cancer and MSTO-211 lung mesothelioma cell cultures was inhibited through the upregulated expression of miRNAs of this family after EGCG treatment [193]. EGCG and quercitin were able to reduce invasiveness of BxPc-3 and MIA-PaCa2 human pancreatic ductal adenocarcinoma cells in culture by upregulating miR-let-7a that belongs to the let-7 miRNA family [194].
Curcumin also modulates miRNA expression. In a recent study, the ability of this polyphenol to induce global changes in mRNA and miRNA expression in MCF-7, MDA-MB-321, and T47D human breast cancer cell cultures was established; the exact pattern of changes in RNA expression was individual for each cell line [195]. Downregulation of oncogenic miR-21 miRNA expression by curcumin suppressed proliferation of human MCF-7 breast cancer [196], AGS gastric adenocarcinoma [197], A549 non-small cell lung adenocarcinoma [198], and Rko and HCT116 colon carcinoma cell cultures [199]. For colon carcinoma cell cultures, a significant decrease in invasiveness and motility was also observed. The ability to inhibit miR-21 expression was demonstrated not only for curcumin, but for its synthetic analogs as well [200][201]. Treatment of MCF-7 human breast cancer cell cultures with this polyphenol downregulated expression of oncogenic miR-19a and miR-19-b, leading to inhibition of proliferation [202]. Curcumin also has the ability to simultaneously modulate the expression of multiple miRNAs: changes in the level of expression of around 30 miRNAs were detected in a BxPC-3 human pancreatic carcinoma cell culture after curcumin treatment [203]. This polyphenol also upregulated four miRNAs and downregulated miR-136 and miR-186* in a A549/DDP human lung adenocarcinoma cell culture, thus leading to induction of apoptosis [204]. The effects of curcumin on expression of miR-34 and miR-98 were also demonstrated: curcumin-mediated increase in the expression of miR-34 led to inhibition of the proliferation of 22RV1, PC-3, and DU145 human prostate carcinoma cell cultures [205] and miR-98 of A549 human lung adenocarcinoma cell culture [206]. In the latter case, treatment with this phenolic compound also decreased invasiveness of tumor cells by downregulating expression of MMP2 and 9 extracellular matrix metalloproteinases.
Modulation of miRNA expression in several cancer cell lines that led to inhibition of the proliferation and induction of apoptosis was also demonstrated for quercitin. Treatment with this polyphenol upregulated expression of miR-16 in HSC-6 and SSC-9 human oral cavity squamous cell carcinoma cell cultures, leading to inhibition of proliferation [207]; similar downregulation of this miRNA expression in A549 human lung adenocarcinoma cell culture by this polyphenol led to a decrease in claudin-2 expression [208]. Suppression of the proliferation and induction of apoptosis by quercitin-induced upregulation of the tumor suppressor miR-34 in HepG2 human hepatocellular carcinoma cell culture was also demonstrated [209]. It was established that treatment with this plant phenolic compound upregulates expression of miR-146a in MCF-7 and MDA-MB-231 human breast cancer cell cultures and mouse xenograft models, leading to inhibition of proliferation [210]. Treatment of BEAS-2B normal human bronchial epithelial cells with quercitin prevented induction of their Cr(VI) ions-induced malignant transformation by downregulation of miR-21 expression, both in cell cultures and in the mouse xenograft model [211]. Quercitin was also able to suppress the proliferative activity of several pancreatic ductal carcinoma cell lines in culture and in xenograft models by upregulating let-7c miRNA expression [212]. In combination with resveratrol, this polyphenol exerted antiproliferative and proapoptotic activities towards HT-29 human colon adenocarcinoma cell culture by downregulating miR-27a [213].
Resveratrol also causes modulatory activity towards miRNA expression in tumor cells. It was demonstrated that treatment of murine C6 glioma cell cultures and xenografts with this phenolic compound downregulated expression of miR-21, miR-30a-5p, and miR19, leading to global changes in cellular proteome and inhibition of proliferative activity [214]. Antiproliferative and proapoptotic activity of resveratrol mediated by downregulation of miR-21 was shown for human U251 glioblastoma [215], T24 and 5637 urinary bladder carcinoma [216], and DU145 prostate carcinoma cell cultures [217]. Treatment of human HT-29 and HCT-116 colon cancer and U87 and U251 malignant glioma cell cultures with resveratrol resulted in a reduction in their invasiveness and motility mediated by upregulation of mir-34c in colon carcinoma and miR-34a in malignant glioma cells [218][219]. In MCF-7 and MDA-MB-231 breast cancer cell cultures, this phenolic compound was able to promote apoptosis by modulating expression of more than 40 miRNAs; miR-542-3p, miR-122-5p, miR-199a-5p, miR-125b-1-3p, miR-140-5p, and miR-20a-5p, known for participation in tumorigenesis, were the most important among them [220].
Genistein also has modulatory activity towards miRNA expression. Treatment of human C918 uveal melanoma C918 [221] and SKOV3 ovarian cancer cell cultures [222] inhibited their proliferation by downregulating miR-27a expression. Remarkably, genistein-induced decrease in the viability of cultivated human A549 non-small cell lung adenocarcinoma cells was mediated by the opposite effect: expression of miR-27a was upregulated by polyphenol treatment [223]. Suppression of the proliferation of human MCF-7 breast cancer cell culture by genistein treatment-induced upregulation of miR-23b expression was demonstrated [224]. This plant polyphenol also downregulated expression of miR-151 in human DU145 and PC3 prostate cancer cell cultures, thus suppressing cellular mobility and invasiveness [225]. Downregulation of miR-155 by genistein has a proapoptotic effect on human MDA-MB-435 aggressively metastasizing breast cancer cell cultures [226]. This phenolic compound also has antiproliferative and proapoptotic effects on human AsPC-1 and MiaPaCa-2 pancreatic cancer [227] and PC3 и DU145 prostate cancer cell lines [228] through upregulation of miR-34a expression. It is noteworthy that in the latter case, HOTAIR long regulatory ncRNA, which is associated with malignant cell proliferation and invasiveness, was one of the molecular targets of miR-34a. During the last few years, a growing number of reports on the activity of plant phenolic compounds towards this class of regulatory ncRNAs appeared [229].
A significant amount of data on the modulatory activity of natural polyphenols and on multiple mechanisms of epigenetic regulation were accumulated to the present day, allowing for the possible development of anticancer and chemopreventive drugs that may utilize this activity to prevent or reverse cancer-associated epigenetic disorders. The list of plant polyphenolic compounds that are known to modulate epigenetic regulation systems is not limited to the compounds described above, and is expanding rapidly [230]. The ability of a single polyphenol to simultaneously affect a number of molecular targets may allow a hypothetical polyphenol-based anticancer drug to affect a broad spectrum of tumors and/or alleviate multiple pre-malignant disorders. At the same time, this potential advantage significantly complicates development and application of such hypothetic drugs, as the effect of a given polyphenol on different epigenetic regulation systems might be highly context-dependent and difficult to predict. The mechanisms behind the effects of plant polyphenols on epigenetic regulatory mechanisms are summarized in Table 2.
Table 2. Mechanisms behind the effects of plant polyphenols on epigenetic regulatory mechanisms.
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin | 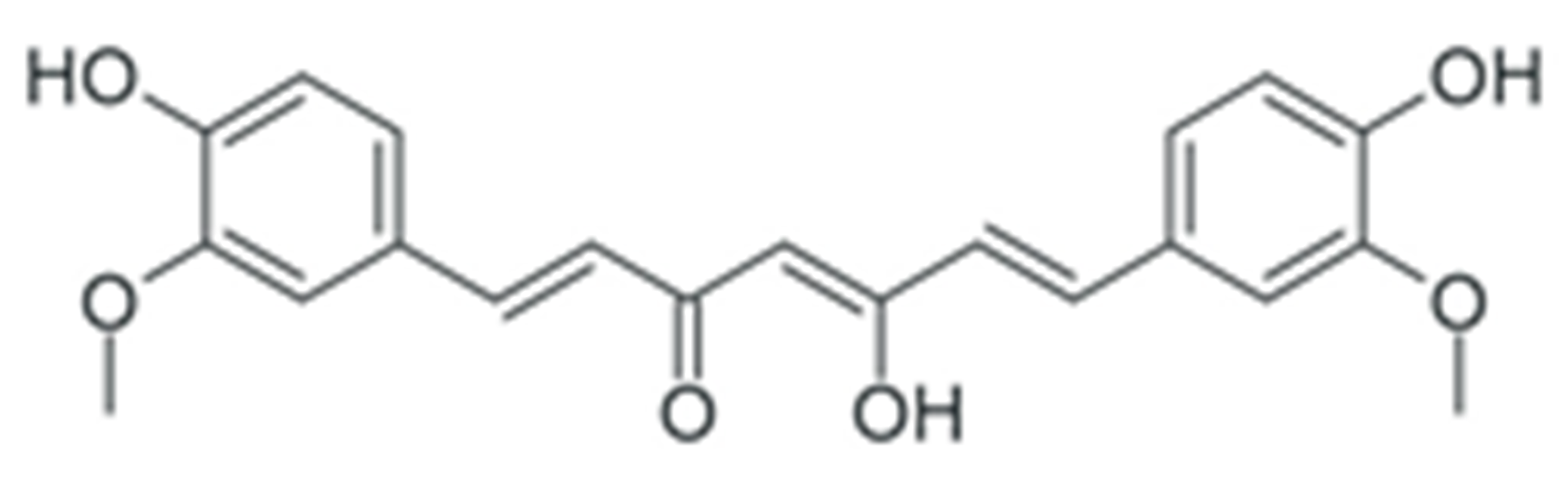 |
Downregulation of DNMT DNA methyltransferases expression | [163][164] |
| Downregulation of histone deacetylase expression | [176][177] | ||
| Modulation of various miRNAs expression | [195] | ||
| Downregulation of miR-21 expression | [196][197][198][199][200][201] | ||
| Downregulation of miR-19a and -19b | [202] | ||
| Modulation of various miRNAs expression | [203] | ||
| Downregulation of miR-136 and -186* | [204] | ||
| Upregulation of miR-34 | [205] | ||
| Upregulation of miR-98 | [206] | ||
| EGCG | 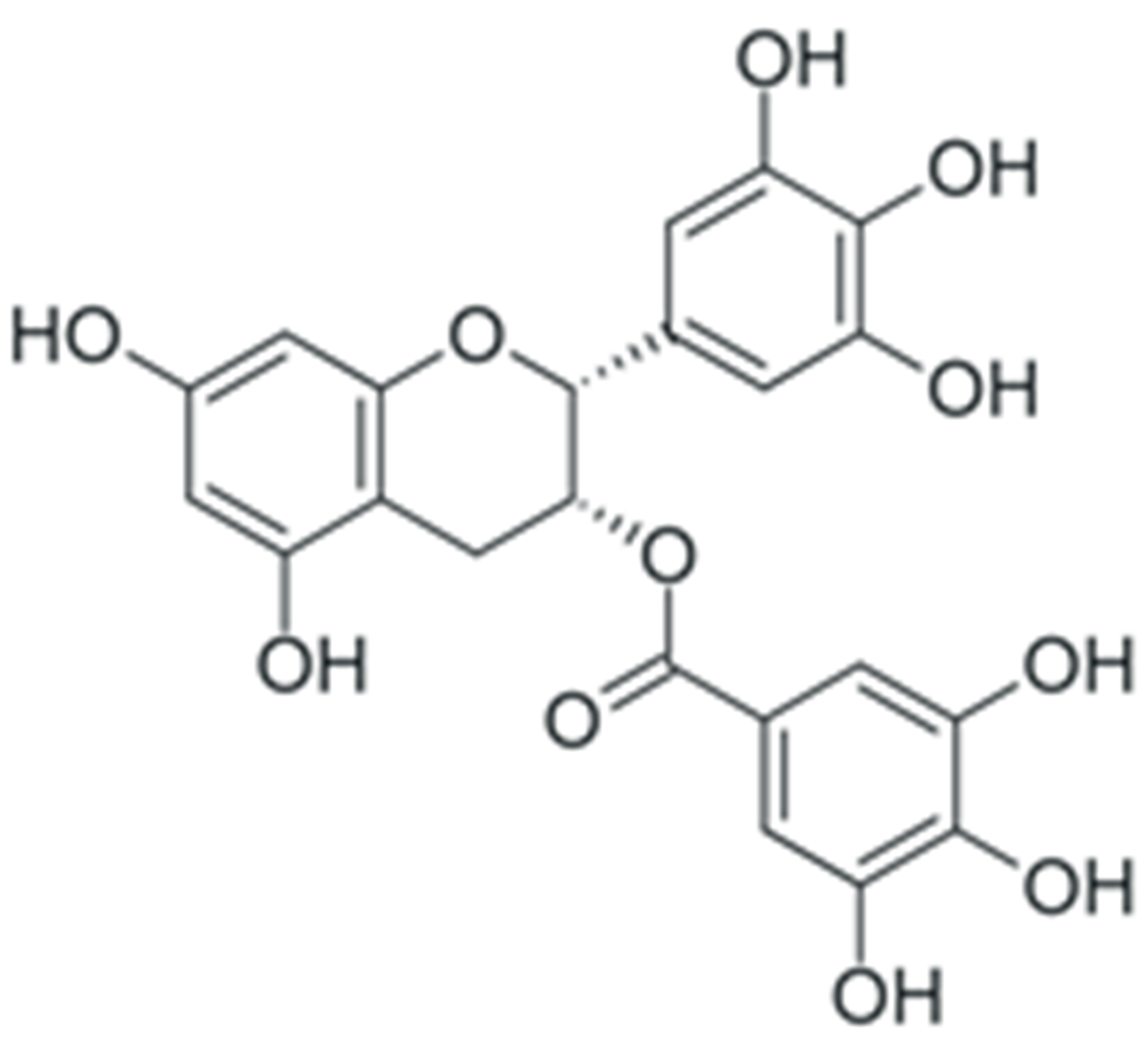 |
Downregulation of DNMT DNA methyltransferases expression | [159] |
| Inhibition of DNMT catalytic activity | [160] | ||
| Inhibition of HDAC 1-3 histone deacetylase expression | [173][174] | ||
| Inhibition of histone acetyltransferase catalytic activity | [175] | ||
| Modulation of various miRNAs expression, including miR-16 | [189] | ||
| Downregulation of miR-92, -93 and -106b, upregulation of miR-7-1, -34a and -99a | [190] | ||
| Upregulation of miR-210 expression | [191] | ||
| Downregulation of miR-25 expression | [192] | ||
| Upregulation of let-7 family miRNAs expression | [193] | ||
| Upregulation of miR-let7a (in combination with quercetin) | [194] | ||
| Quercetin | 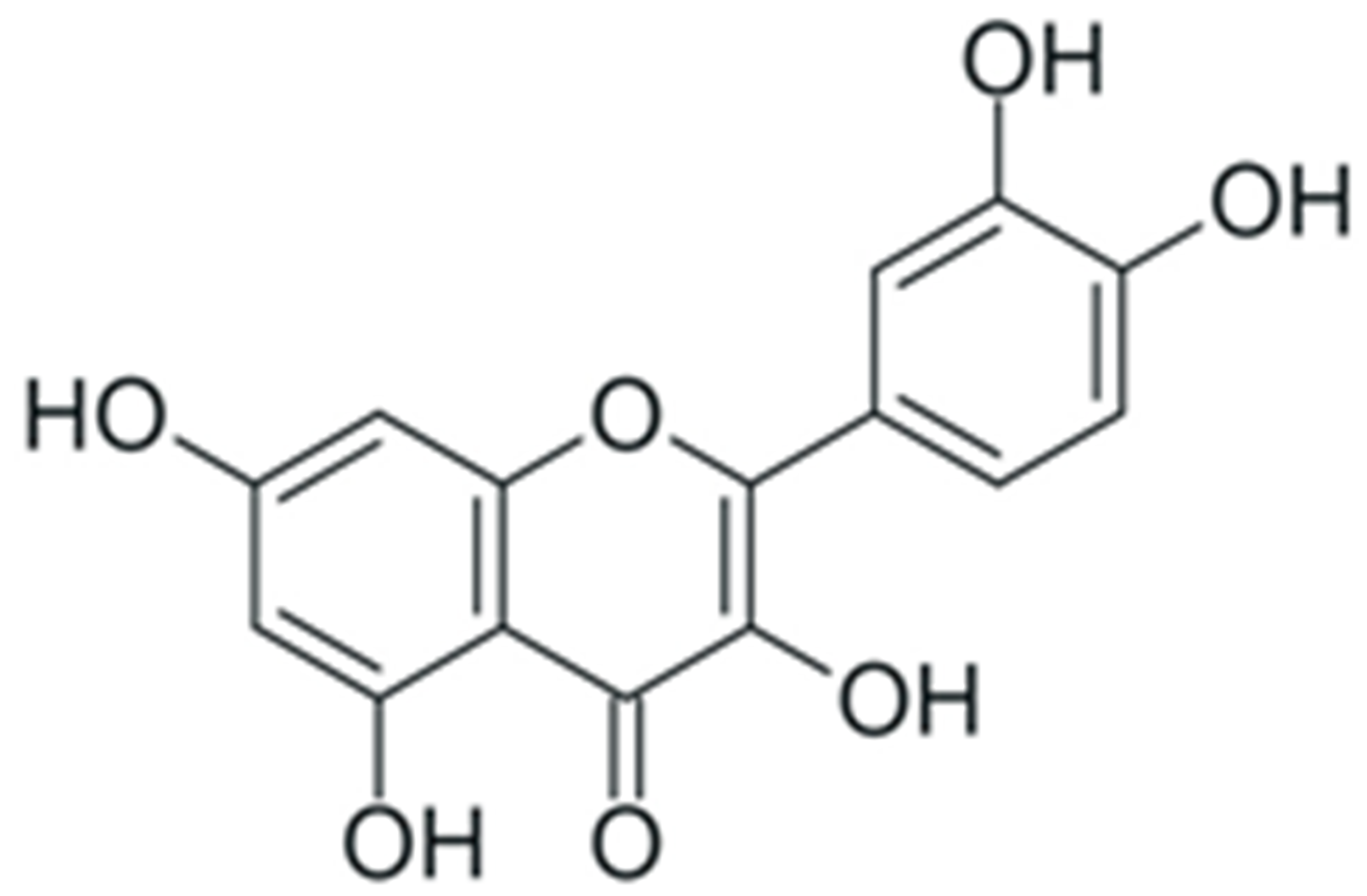 |
Inhibition of DNMT catalytic activity | [166] |
| Inhibition of histone deacetylase and increase in histone acetyltransferase activity | [178] | ||
| Inhibition of p300 acetyltransferase activity | [179] | ||
| Upregulation of miR-16 | [207] | ||
| Downregulation of miR-16 | [208] | ||
| Upregulation of miR-34 | [209] | ||
| Upregulation of miR-146a | [210] | ||
| Downregulation of miR-21 | [211] | ||
| Upregulation of let-7c | [212] | ||
| Downregulation of miR-27a (in combination with resveratrol) | [213] | ||
| Resveratrol | 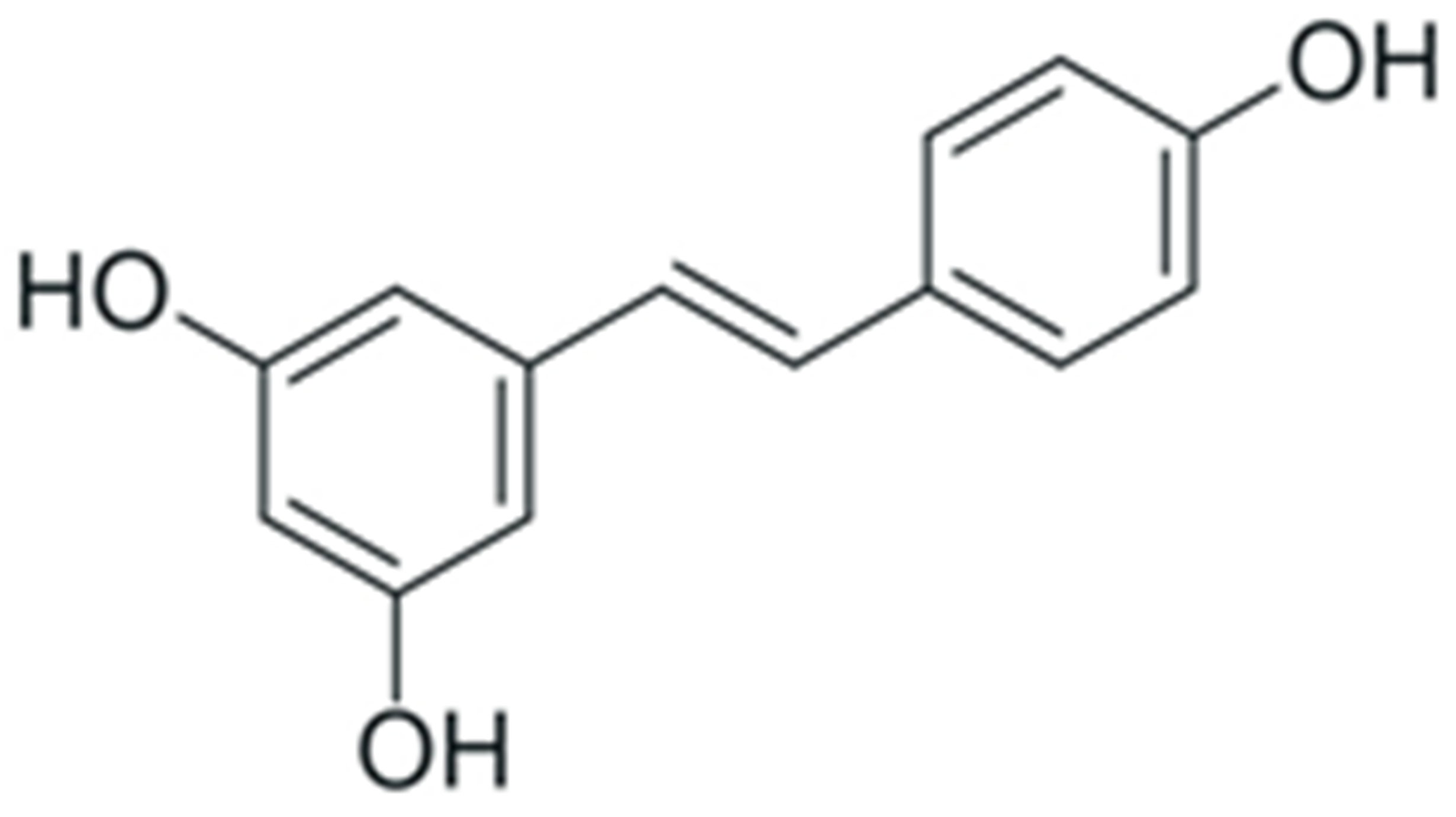 |
Inhibition of DNA methyltransferase recruitment by SIRT1 | [167] |
| Selective reversion of hypermethylation of tumor suppressor genes (in combination with pterostilbene) | [168][169] | ||
| Recruitment of DNMT3b DNA methyltransferase to oncogenes | [168] | ||
| Enhancement of SIRT1 histone deacetylase activity | [180] | ||
| Modulation of NuRD histone deacetylase complex activity | [181][182] | ||
| Downregulation of miR-19, -21 and -30-a5p | [214] | ||
| Downregulation of miR-21 | [215][216][217] | ||
| Upregulation of miR-34a and -34c | [218][219] | ||
| Modulation of various miRNAs expression, including miR-20a-5p, -140-5p, -125b-1-3p, -199a-5p, -122-5p and -542-3p | [220] | ||
| Genistein | 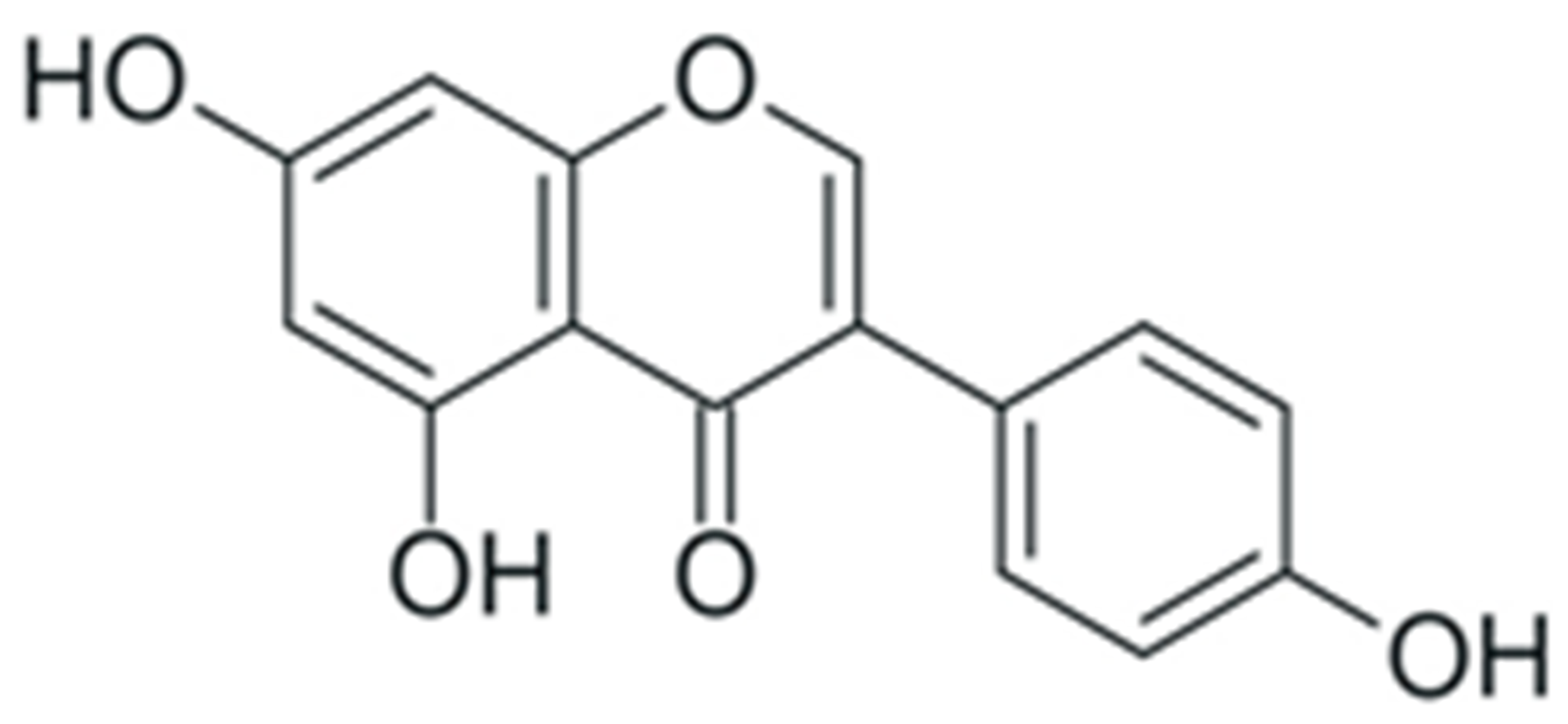 |
Inhibition of DNMT catalytic activity | [170][171][172] |
| Promotion of histone acetylation at regulatory regions of tumor suppressor genes | [170][171][183][184] | ||
| Downregulation of miR-27a | [221][222] | ||
| Upregulation of miR-27a | [223] | ||
| Upregulation of miR-23b | [224] | ||
| Downregulation of miR-151 | [225] | ||
| Downregulation of miR-155 | [226] | ||
| Upregulation of miR-34a | [227][228] |
5. Tumor-Promoting Inflammation
Inflammation is a complex response of innate and acquired components of the immune system after a traumatic or infective injury of tissues that includes activation, recruitment, and proliferation of various immune cells and facilitates tissue regeneration and restoration of its normal functioning. The exact characteristics of a site of inflammation, such as composition of immune cells attracted to it and chemical factors secreted by these cells, are dependent on the nature of the damage that caused the inflammatory response. Acute inflammation usually resolves completely after the inflammation-causing lesion is repaired, but in certain cases, it may switch to a chronic inflammation, or in the case of systemic inflammation, which is not caused by infection or trauma, it might develop as a chronic form. A site of chronic inflammation can be viewed as “a wound that never heals” [231]. Damage repair at the inflammatory site must include elimination of tissue debris, modification of the extracellular matrix, induction of angiogenesis, and survival and proliferation of normal cells. These processes facilitate the return of a damaged tissue to homeostasis, but they may also promote malignant transformation: certain forms of immune response are carried out by the production of DNA-damaging reactive oxygen and nitrogen species by neutrophils and macrophages, the induction of angiogenesis supplies the growing tumor with nutrients and oxygen, and modifications of extracellular matrix facilitate the process of metastasizing [232]. These events mark chronic inflammation as a cancer-enabling characteristic, and anti-inflammatory measures are now considered as an important part of antitumor therapy and chemoprevention.
Transcription factors of the NF-κB family are one of the most important regulators of inflammation-associated gene expression. Each of these factors have the Rel homology domain, which is responsible for dimerization of NF-κB proteins and their binding to DNA [233]. Inactive NF-κB dimers form ternary complexes with IκB inhibitor proteins; ubiquitination and subsequent proteasomal degradation of IκB are necessary steps in NF-κB activation. The canonical pathway of NF-κB activation involves phosphorylation of IKK1/2 IκB kinases mediated by NEMO (NF-κB essential modulator) scaffold protein bound to the kinases. This NF-κB activation pathway can be activated by the signals from toll-like receptors (TLR), interleukin-1 receptors (IL-1R), and tumor necrosis factor receptors (TNFR) [234]. The non-canonical NF-κB activation pathway is independent of NEMO activity and is activated by signals from the B-cell activating factor receptor (BAFFR), lymphotoxin β receptor (LTβR), receptor activator of NF-κB (RANK), TNFR2, and fn14. Activation of these receptors leads to activation of NF-κB-inducing kinase (NIK) that phosphorylates IKK1. Activated IKK1 phosphorylates the p100 NF-κB precursor protein, inducing its processing and thus leading to an increase in the concentration of active NF-κB p52 in cytoplasm [235]. IKK1 also phosphorylates those p100 molecules that are bound to preexisting NF-κB dimers, thus acting as their inhibitors (inhibitory p100 is known as IκBδ). Phosphorylation of IκBδ leads to its dissociation and to activation of the NF-κB dimer it was bound to. Canonical and non-canonical pathways of NF-κB activation differ by the intensity and duration of NF-κB activation and by genes regulated by these pathways: the canonical pathway is mainly responsible for activation of an inflammation-associated response, while the non-canonical pathway is involved in cell differentiation and organogenesis [236]. Activation of NF-κB stimulates the cytotoxic immune cell-mediated antitumor activity [237], and also induces expression of antiapoptotic genes, pro-inflammatory cytokines (TNF-αandIL-1, 6 and 8) [238], angiogenesis factors (VEGFand its receptors) [239], and cell metabolism regulators (HIF-1α) [240], thus facilitating the malignant transformation.
Plant phenolic compounds might inhibit NF-κB activation at different stages of signal transduction (Figure 3). Treatment of HCA-7 human colon adenocarcinoma cell culture with EGCG led to the general decrease in NF-κB activity [241]. This phenolic compound was also able to inhibit IKK activity in A549 human lung cancer cell culture, preventing the activation of NF-κB by TNF-α [242]. EGCG also prevented IL-1β-mediated degradation of IRAK receptor-associated kinase in this cell culture, thus inhibiting IKK activation, IκBα degradation, and NF-κBp65 phosphorylation [243]. Inhibition of IKK activity by this phenolic compound was also demonstrated for rat IEC-6 normal small intestine epithelial cell culture [244]. Treatment of rheumatoid arthritis-associated synovial fibroblast culture with EGCG prevented activation of IKK by inhibiting TAK-1 TNF-β-dependent kinase [245][246]. Molecular modeling studies of EGCG interaction with components of NF-κB signal transduction pathways suggested that the inhibitory activity of this compound may be mediated by its interaction with ATP binding site ofTAK-1 and IRAK kinases. The inhibitory activity of this plant phenolic compound towards NF-κB signaling pathway was also demonstrated in vivo: treatment with EGCG alleviated the development of picrylsulfonic acid-induced colitis in Wistar rats by inhibiting activation of this transcription factor, presumably by binding to the IKK inhibitor binding site, as suggested by the results of molecular docking [247]. The ability of EGCG to inhibit the interaction of NF-κB with DNA by covalent modification of thiol groups of cysteine residues of this protein was also demonstrated [248].
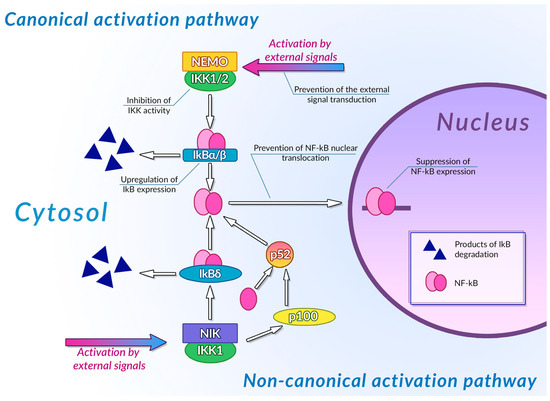
Figure 3. Inhibition of NF-κB signaling pathways by plant polyphenols. See text for detail.
The inhibitory activity of curcumin towards NF-κB signaling pathway was also demonstrated. Treatment of MDA-MB-231 human breast cancer cell cultures with this phenolic compound inhibited NF-κB-mediated expression of CXCL-1 and -2 pro-inflammatory cytokines by preventing IκB phosphorylation [249]. In RAW264.7 murine macrophage cultures, curcumin was able to reduce the efficiency of NF-κB binding to DNA, thus leading to inhibition of TNF-α, IL-1β, and IL-6 expression [250]. Although this polyphenol failed to demonstrate similar activity towards MCF-7 human breast adenocarcinoma cell cultures, curcumin-loaded chitosan-protamine nanoparticles did reduce the levels of NF-κB, IL-6 and TNF-α [251]. Treatment of T47D human breast cancer cell culture with curcumin suppressed the activity of NF-κB signaling pathway both by suppressing activity of this transcription factors and by downregulating expression of NF-κB and IKK [252]. Downregulation of NF-κB expression upon curcumin treatment in HeLa human cervical carcinoma cell culture was also observed [253]. Treatment of Eca109 and EC9706 esophageal squamous cell carcinoma cell cultures and xenografts with this plant polyphenol also resulted in downregulation of NF-κB expression, as well as in suppression of IκB phosphorylation [254]. In WERI-Rb-1 human retinoblastoma cell culture, curcumin suppressed the activation of NF-κB, thus preventing its nuclear translocation and downregulating expression of VEGF and MMP-2 and -9 [255].
Quercitin has activity towards NF-κB and its regulators. Treatment of Caco2 human colon adenocarcinoma cell culture with this phenolic compound downregulated expression of NF-κBp65, thus suppressing cellular motility and invasiveness [256]. Expression of the other components of the NF-κB signaling pathway was also affected by quercitin. In Caco-2 and SW-620 human colon carcinoma cell cultures, this polyphenol demonstrated pro-apoptotic activity by upregulating expression of IκBα, suppressing its phosphorylation, and reduction in the efficiency of NF-κB binding to DNA [257]. It was also established that in H460 non-small cell lung cancer cell cultures, quercitin was able to promote apoptosis by downregulating expression of NF-κB while simultaneously upregulating expression of IκBα [258]. Treatment of SAS human oral squamous cell carcinoma cell culture with this phenolic compound resulted in suppression of cellular invasiveness by simultaneously reducing both NF-κB and IκB expression and lowering the presence of phosphorylated IKK1/2 [259]. Quercitin also exerted its activity towards NF-κB during in vivo studies: treatment with this compound abrogated induction of oral adenocarcinoma in Syrian hamster buccal pouches with dimethylbenzanthracene by promoting apoptosis of tumor cells and downregulating expression of NF-κBp50 and p65 [260]. Similar results were obtained during induction of tumorigenesis by dimethylhydrazine in rats: treatment with quercitin lowered the number of colonic tumors and prevented the upregulation of NF-κB expression [261].
A certain degree of controversy exists regarding the modulatory activity of resveratrol towards the NF-κB signaling pathway. It was established that treatment of several human cervical cancer cell cultures with this phenolic compound resulted in reduction in the proliferation and induction of apoptosis; for CaSki cell cultures, these effects were the most prominent. In C33A, HeLa, CaLo, and CaSki cell cultures, the antiproliferative and proapoptotic activities of resveratrol were accompanied by a significant decrease in NF-κB p65 expression [262]. In human colon cancer cell cultures, resveratrol prevented nuclear translocation of NF-κB, presumably by binding to its monomer, and thus preventing its dimerization [263]. This phenolic compound was also able to suppress proliferation of human MV3 and A375 melanoma cells, both in culture and in a mouse xenograft model, by downregulating the expression of NF-κB and NF-κB-regulated miR-221, thus leading to the enhancement of the expression of TFG tumor suppressors, the mRNA of which is targeted by miR-221 [264]. The chemopreventive activity of resveratrol, mediated by inhibition of NF-κB activity with this polyphenol, was also reported. Treatment of KC mice that spontaneously develop pancreatic dysplasias and malignant tumors with resveratrol led to reduction in the number and severity of pre-malignant lesions and to suppression of NF-κB activity in these structures [265]. After induction of hepatocelular carcinoma in rats by ethanol and aflatoxin treatment, this plant phenolic compound alleviated the induction of malignant transformation by preventing the reduction in the activity of antioxidant enzymes and increasing the NF-κB-inhibiting SIRT1 activity [266]. Nevertheless, treatment with resveratrol did not result in suppression of tumor cell growth. This polyphenol was able to suppress proliferation of NF-κB-overexpressing SKOV3 human ovarian cancer cells in aggregates by inhibiting activation of this transcription factor. At the same time, resveratrol failed to demonstrate any significant antiproliferative activity towards OVCAR5 ovarian cancer cell aggregates, whose expression of NF-κB is far less pronounced [267]. Treatment with resveratrol also had no effect on the secretion of VEGF by OVCAR5 cells, while treatment of SKOV3 aggregates with this phenolic compound resulted in a significant decrease in the efficiency of the secretion of this factor. Moreover, secretion of pro-survival interleukin IL-8 by both cell cultures increased significantly upon treatment with resveratrol. Growth stimulatory activity of low concentrations (less than 10 µM) of resveratrol towards MDA-MB-495c human breast cancer cell culture was reported; at the same time, this polyphenol has antiproliferative activity towards MDA-MB-231 and MCF-7 human breast cancer and DU145 human prostate adenocarcinoma cell cultures in all concentration values [268]. The stimulatory effect of resveratrol treatment on MDA-MB-495c cell culture was exerted by promotion of IκB phosphorylation and NF-κB expression, and by facilitation of nuclear translocation of NF-κB.
Certain cases of genistein activity towards the NF-κB signaling pathway are also controversial. Genistein causes pro-apoptotic activity towards U266 human multiple myeloma cell culture by upregulating miR-29b that suppresses NF-κB p65 expression [269]. It was established that treatment of CAL-62 and CGTH-W1 human thyroid carcinoma cell cultures with this phenolic compound resulted in a decrease in the viability of cultured cells through a reduction in the amount of mRNAs of several pro-tumorigenic proteins, including NF-κB [270]. Reduction in the amount of NF-κB mRNA after genistein treatment was also detected for A549 human lung carcinoma, along with pro-apoptotic activity and downregulation of AKT, HIF1, and COX-2 expression [271]. In HT-29 human colon carcinoma cell culture, this polyphenol promoted apoptosis and reverted the epithelial-to-mesenchymal phenotype transition by downregulating expression of NF-κB and Notch-1, thus causing a decrease in expression of invasiveness-related proteins and upregulation of the expression of pro-apoptotic factors [272]. Results of treatment of MDA-MB-231 human breast cancer cell culture with genistein also suggest involvement of Notch-1 in the inhibition of the NF-κB signaling pathway by this phenolic compound, as genistein exerted antiproliferative and proapoptotic activity towards this cell culture by downregulating expression of Notch-1, accompanied by inhibition of NF-κB activation [273]. Genistein was also able to suppress proliferation of LoVo and HT-29 human colon carcinoma cell cultures by preventing IκBα phosphorylation, thus abrogating phosphorylation of NF-κB p65 and its nuclear translocation [274]. It is noteworthy that genistein- and daidzein-rich biotransformed soybean extract has pro-apoptotic activity towards A375 human melanoma cell culture by the opposite mechanism: overexpression of TNF receptors TNFR1/2 caused by this treatment led to IKK activation and to an increase in NF-κBp65 phosphorylation [275]. Moreover, treatment of HT-29 and SW620 human colon adenocarcinoma cell cultures with genistein decreased the viability of the latter culture [276]. This decrease in viability, however, was induced by an increase in H2O2 concentration caused by the upregulation of the expression of antioxidant enzymes, such as SOD1 and 2, accompanied by activation of NF-κB nuclear translocation and subsequent stimulation of the expression of pro-inflammatory proteins, such as TNF and CXC chemokines. These effects were observed for both cell cultures, but in HT-29 cell culture, they had no significant proapoptotic effect.
The anticancer and chemopreventive activity of plant polyphenols, carried out through activation of the NF-κB signaling pathway by these compounds, was demonstrated on a multitude of objects, including cell cultures, tumor xenografts, and the models of induced carcinogenesis. Suppression of NF-κB activation led to a decrease in the efficiency of antiapoptotic, pro-angiogenic, and prometastatic inflammatory signals in tumor tissue. Mechanisms of modulation of the NF-κB signaling pathway by plant polyphenols are summarized in Table 3. The anti-inflammatory activity of natural phenolic compounds towards normal tissues was also demonstrated, and the mechanisms underlying this activity were not limited to inhibition of NF-κB activation [277]. These mechanisms may also include inhibition of activity of the other key pro-inflammatory enzymes and signaling molecules, such as COX-2 cyclooxygenase [278], iNOS inducible NO synthase275, and pro-inflammatory cytokines [279]. These properties suggest the possible use of polyphenols as chemopreventive drugs that suppress pro-tumorigenic influence of inflammation during the entire tumorigenesis. Nevertheless, evidence of growth-stimulatory and pro-inflammatory activity of natural phenolic compounds in cell cultures calls out for the additional study of the activity of polyphenols in the inflammation sites before such drugs could be designed and introduced in clinical practice.
Table 3. Modulation of NF-κB signal transduction pathway by plant polyphenols.
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Curcumin | 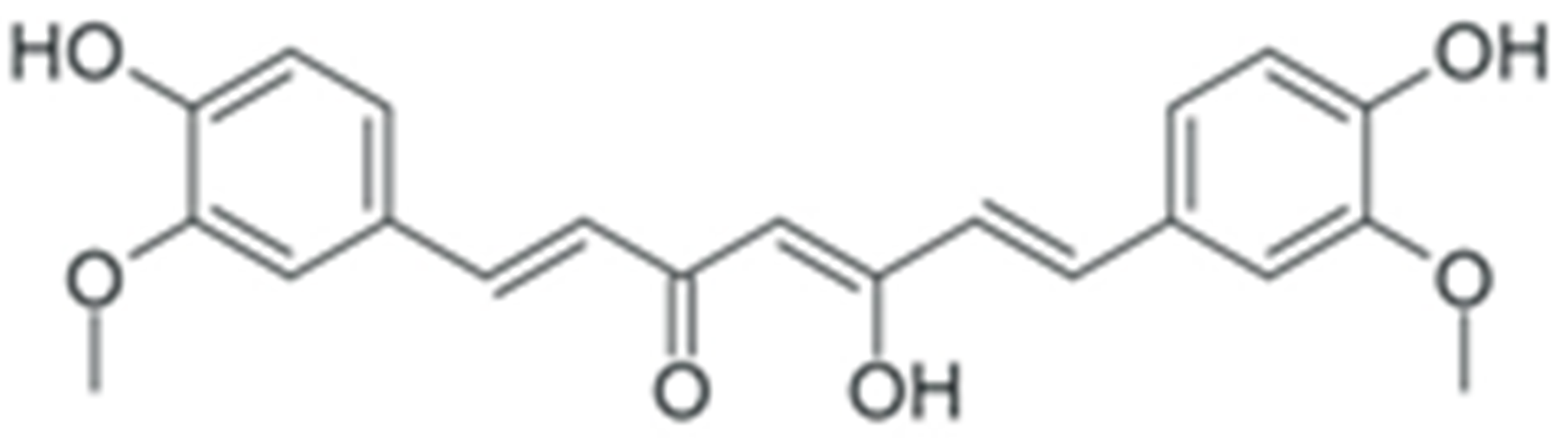 |
Prevention of IκB phosphorylation | [249] |
| Reduction in NF-κB DNA binding efficiency | [250] | ||
| Reduction in NF-κB level | [251] | ||
| Downregulation of NF-κB and IKK expression | [252] | ||
| Downregulation of NF-κB expression | [253] | ||
| Downregulation of NF-κB and suppression of IκB phosphorylation | [254] | ||
| Prevention of NF-κB activation and nuclear translocation | [255] | ||
| EGCG | 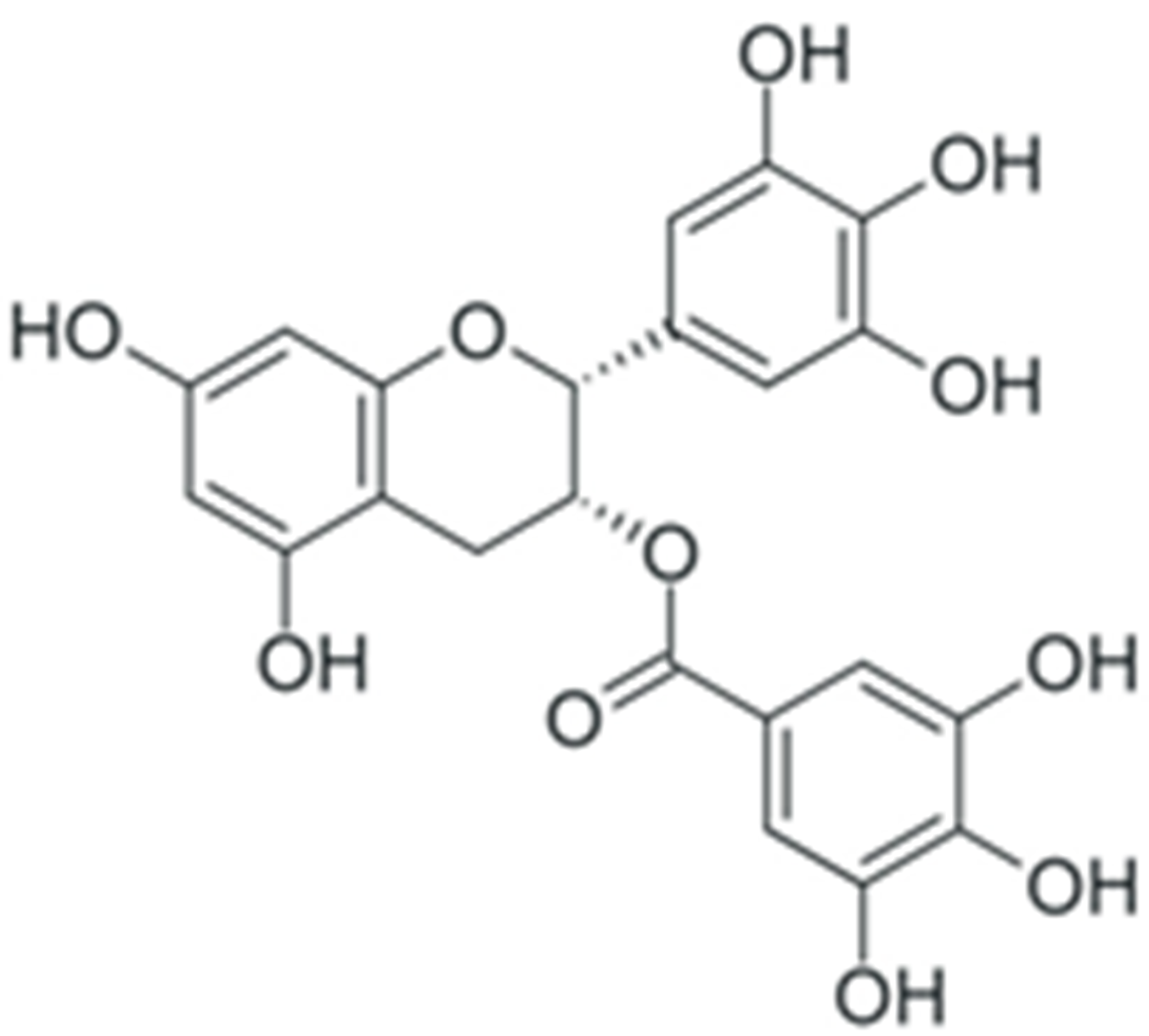 |
General decrease in NF-κB activity | [241] |
| Inhibition of IKK activity | [242][244] | ||
| Prevention of IRAK receptor-asociated kinase degradation | [243] | ||
| Inhibition of TAK-1 TNF-β-dependent kinase | [245][246] | ||
| Inhibition of NF-κB activity by binding to IKK inhibitor binding site | [247] | ||
| Prevention of NF-κB interaction with DNA | [248] | ||
| Quercetin | 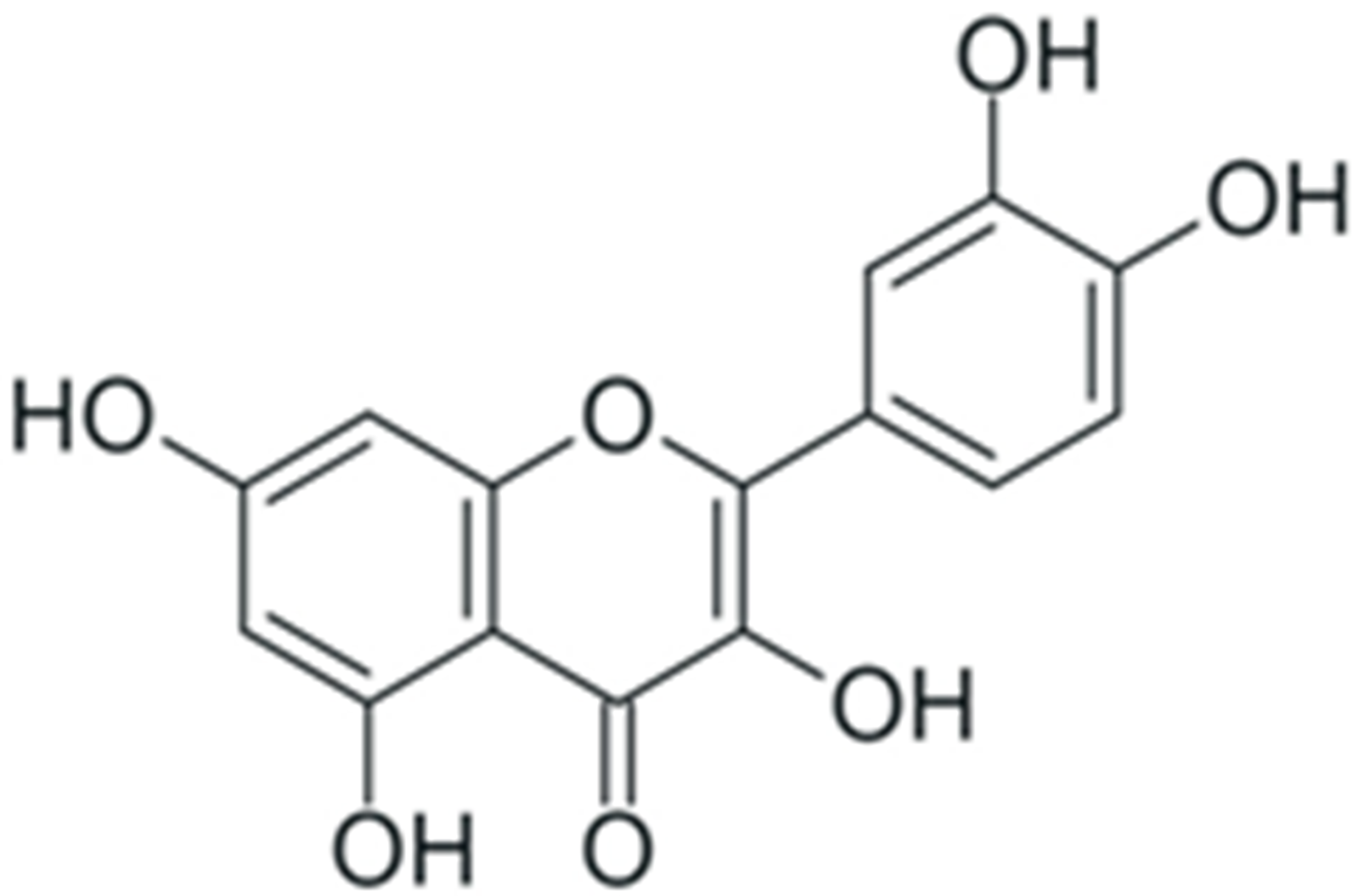 |
Downregulation of NF-κB p65 expression | [256] |
| Upregulation of IκBα expression | [257] | ||
| Downregulation of NF-κB expression, combined with upregulation of IκB expression | [258] | ||
| Downregulation of NF-κB and IκB expression, reduction in IKK1/2 phosphorylation | [259] | ||
| Downregulation of NF-κB p65 and p50 expression | [260] | ||
| Prevention of upregulation of NF-κB expression | [261] | ||
| Resveratrol | 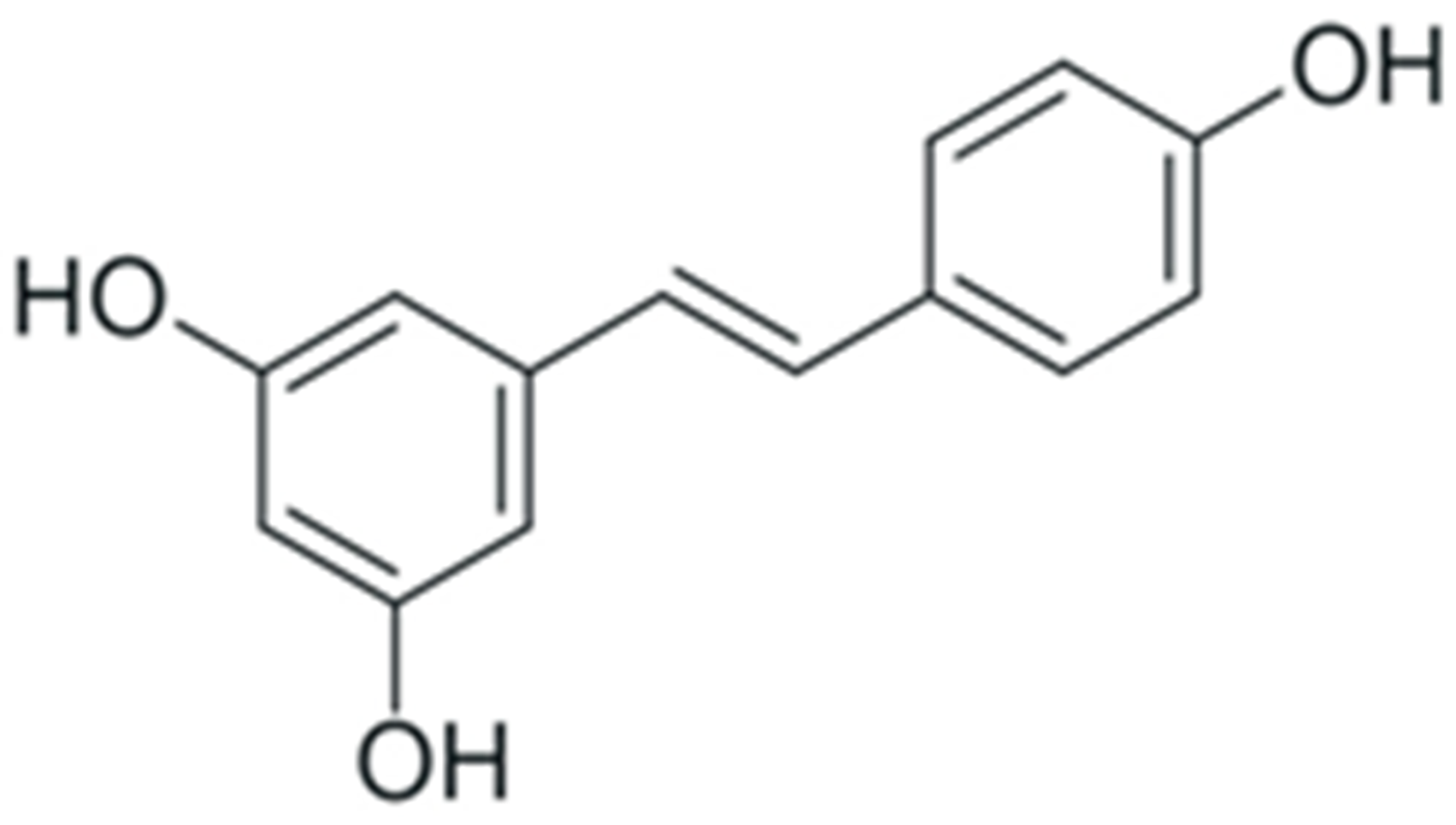 |
Decrease in NF-κB p65 expression | [262] |
| Prevention ov NF-κB dimerization and nuclear translocation | [263] | ||
| Downregulation of NF-κB expression | [264] | ||
| Suppression of NF-κB activity | [265] | ||
| Promotion of NF-κB-inhibiting activity of SIRT1 | [266] | ||
| Inhibition of NF-κB activation, combined with an increased secretion of IL-8 | [267] | ||
| Promotion of NF-κB expression and nuclear translocation, stimulation of IκB phosphorylation | [268] | ||
| Genistein | 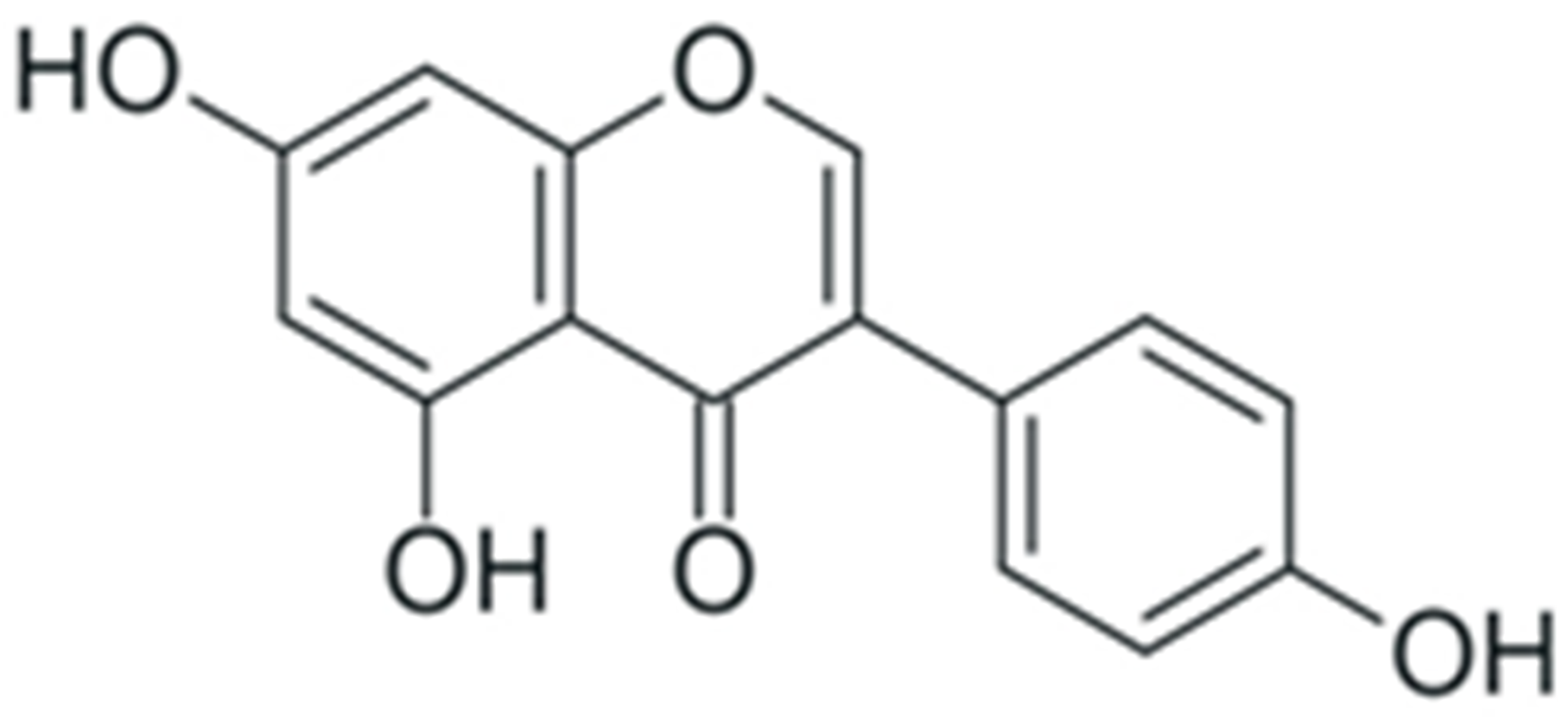 |
Suppression of NF-κB expression by upregulation of miR-29 | [269] |
| Downregulation of NF-κB expression | [270][271] | ||
| Downregulation of NF-κB expression by suppression of Notch-1 activity | [272][273] | ||
| Inhibition of IκB and NF-κB phosphorylation, prevention of NF-κB nuclear translocation | [274] | ||
| Activation of IKK, increase in NF-κB phosphorylation (in combination with daidzein) | [275] | ||
| Facilitation of NF-κB activation and nuclear translocation | [276] |
6. Interactions with Cancer-Associated Microbiota
Human organisms are inhabited by microorganisms that modulate normal physiological processes [280] and various diseases [281], including malignant tumors [282]. Certain microorganisms of human microbiota might produce genotoxic substances capable of damaging the host cellular genome; colibactin-producing Escherichia coli [283] and cytolethal distending toxin-producing Campylobacter jejuni [284] are the examples of such microorganisms. Other bacterial toxins may promote tumorigenesis by modulation of cellular signaling pathways: toxins produced by F. nucleatum activate the β-catenin signaling pathway and suppress the antitumor activity of NK cells [285]. CagA and VacA proteins secreted by H. pylori attenuate activity of tumor suppressors, stimulate proliferation, facilitate the acquisition of the mesenchymal phenotype, and modulate the host immune response [286]. Accumulated evidence of procarcinogenic activity of this microorganism was sufficient to classify it as a IARC class 1 carcinogen [287]. Non-specific products of bacterial metabolism might also affect the process of tumorigenesis. Bacterial metabolites of fatty acids, trimethylamine-n-oxide, and hydrogen sulfide facilitate development of tumors, whereas short-chain fatty acids (SCFA) and niacin suppress this process [288]. It is also established that colonic malignancies are often accompanied by changes in the species composition of gut microbiota [289], but it is largely unclear whether these changes are the cause of tumorigenesis or the consequence of this process [290].
Plant polyphenols have antibacterial properties [291], which allow them to modulate the intestinal microflora. Silibinin was able to suppress the H. pylori culture growth by promoting changes in bacterial morphology and suppressing cell division [292]. Kaempferol and (-)-epicatechin demonstrated antibacterial and bacteriostatic effects towards this pathogen in vitro [293]. Similar activity was demonstrated for ellagic and gallic acid, as well as for quercitin aglycon and its glycosylated form [294]. Antibacterial activity of baicalin and baicalein towards this microorganism was registered both in vitro and in vivo: these phenolic substances decreased the bacterial load in H. pylori-infected mice by inhibiting the adhesiveness and invasiveness of this microorganism [295]. Antibacterial activities of polyphenol-containing natural and artificial mixtures were also detected. Citrus juices and extracts from citrus plants have antibacterial activity towards H. pylori culture in vitro, and alleviated H. pylori-induced gastritis in vivo by suppressing bacterial colonization of mucosa and abrogating inflammation elicited by the infection [296]. Similar antibacterial activity of extracts of fruits of Rubus crataegifolius and bark of Ulmus macrocarpa was described [297]. The antibacterial activity of plant polyphenols towards H. pylori was determined by the ability of these substances to impair functioning of transmembrane energetic of this microorganism [298].
Natural phenolic compounds demonstrated antibacterial properties towards C. jejuni as well. The antibacterial activity of curcumin was demonstrated by treatment of C. jejuni culture with this substance [299]. Similar antibacterial properties of EGCG were also demonstrated, along with the ability of this polyphenol to impair motility and biofilm formation by these microorganisms and to suppress activity of bacterial autoinducer-2, thus disturbing quorum-sensing activity [300]. Treatment with resveratrol prevents C. jejuni-induced loosening of cellular junctions between intestinal epithelial cells and alleviates infection-induced apoptosis both in vitro and in vivo, thus facilitating epithelial barrier function and preventing development of inflammation [301][302].
EGCG, theaflavines and other tea polyphenols have antibacterial activity towards F. nucleatum in vitro by disturbing integrity of the plasma membrane of this organism and by reducing its adhesiveness along with the ability of biofilm formation [303]. Similar activity was demonstrated for complexes of resveratrol with cyclodextrane together with inhibitory activity towards F. nucleatum-induced inflammation [304]. Treatment of female rats on a high-fat diet with genistein prevented the increase in the presence of Enterobacteriaceae in the microflora of their progeny, accompanied by a lower risk of mammary cancer recurrence [305]. The antibacterial activity of plant polyphenols towards cancer-associated microorganisms is summarized in Table 4.
Table 4. Antibacterial activity of plant polyphenols towards cancer-associated microorganisms.
| Compound | Structure | Mechanism | Reference |
|---|---|---|---|
| Silibinin | Flavonol and phenolic acid heterodimer | Suppression of H. pylori growth | [292] |
| Kaempferol | Flavonol | [293] | |
| (-)-epicatechin | Flavanol | ||
| Ellagic acid | Tannin | [294] | |
| Gallic acid | Phenolic acid | ||
| Quercitin | Favonol | ||
| Curcumin | Phenolic acid homodimer | Suppression of C. jejuni growth and prevention of C. jejuni-induced lesions | [299] |
| EGCG | Flavanol and phenolic acid heterodimer | [300] | |
| Resveratrol | Stilbene | [301][302] | |
| EGCG | Flavanol and phenolic acid heterodimer | Suppression of biofilm formation by F. nucleatum, combined with alleviation of inflammation induced by this microorganism | [303] |
| Theaflavines | Condensated flavanols | ||
| Resveratrol | Stilbene | [304] |
Microbiota-associated chemopreventive activity of plant polyphenols may be carried out not only by their antibacterial activity towards certain microorganisms, but also by modulation of species composition of gut microbiota. Dietary supplementation of sausages with powder of dried anthocyanine-rich berries reduced the number of intestinal tumors induced by azoxymethane treatment in rats, and alleviated the presence of pro-inflammatory Bilophila wadsworthia in their microflora [306]. Treatment of IL10−/− mice with curcumin in a similar tumorigenesis induction experiment resulted in a decrease in tumor burden and in prevention of cancer-associated changes in microbiota [307]. Anthocyanine-rich extract from Rubus occidentalis berries was able to reduce the number of tumors and abrogate inflammatory response during azoxymethane-induced carcinogenesis in mice, along with promotion of Neisseria and butyrate-producing bacteria growth and suppression of pathogenic H. pylori, Campylobacter, Bacteroides, and Prevotella [308]. Treatment of rats with polymethoxyflavone mix during induction of carcinogenesis by benzo-a-pyrene prevented changes in species composition of intestinal microbiota by alleviating the increase in the abundance of Sphingobacteriia, Bacilli and Gammaproteobacteria classes, Erysipelotrichales and Lactobacillales orders, and the Parabacteroides family, and by promoting an increase in the abundance of butyrate-producing Ruminococcaceae family [309]. Treatment of rats with isoliquiritigenin during azoxymethane/dextransulfate-induced carcinogenesis resulted in the reduction in tumor incidence by preventing the increase in the abundance of Firmicutes phylum in their microflora, while simultaneously decreasing abundance of Bacteroidetes [310]. This phenolic compound also decreased the presence of opportunistic pathogens, such as Escherichia and Enterococcus, while simultaneously increasing the abundance of butyrate-producing bacteria, such as Bytiricicoccus, Ruminococcus, and Clostridium. Induction of carcinogenesis with a high-fat diet in APCmin/+ mice was abrogated by neohesperidin by inducing the opposite direction of changes of abundance of Firmicutes and Bacteroidetes: this phenolic compound decreased the abundance of Bacteroidetes and increased the abundance of Firmicutes and Proteobacteria [311]. The opposite direction of changes in the relative abundance of Firmicutes and Bacteroidetes in these experiments might suggest that the analysis of cancer-associated microbiota changes at the phylum level may not be informative enough, as only a portion of species of aforementioned phyla are associated with carcinogenesis [312]. Results of the experiments of cancer chemoprevention in azoxymethane-treated mice with EGCG speak in favor of this hypothesis. Treatment with EGCG reduced the severity and number of tumors by preventing the changes in the abundance of different taxa of the Firmicutes phylum: this plant phenolic compound prevented the increase in the abundance of Anaerotruncus, Faecalibacterium and Streptococcus and decrease in the abundance of Clostridiaceae, Lactobacillis and Lachnospiraceae311 [313]. EGCG also promoted an increase in the abundance of Bacteroides and decrease in the abundance of Fusobacterium, Ruminococcus, and Veillonella. Dietary supplementation of mice xenograft with MCA-205 and E0771 murine cancer cells with castalagin-rich Myrciaria dubia berries led to sensibilization of MCA-205 to PD-1 inhibitor therapy, and circumvention of resistance of E0771 to these inhibitors [314]. Castalagin exerted this potentiating effect by promoting the selective enrichment of murine gut microbiota with Alistipes, Blautia, and Ruminococcus.
These data emphasize the interplay between intestinal microbiota, the host organism immune system, and tumor microenvironment, and outline its crucial role in the process of tumorigenesis. Contacts with bacteria and their presence in the human organism since the earliest postnatal period are essential for priming the immune system for response to pathogen infections [315]. The interaction of bacteria and their metabolites with host dendritic cells is necessary for developing different immune reactions to commensal vs. pathogenic microorganisms [316]. Activity of intestinal mucosa-associated T lymphocytes is also modulated by microbiota: presence of microorganisms facilitate development CD4+ and cytotoxic CD8+ T lymphocytes [317]. Induction of peripheral RORγ+ regulatory T lymphocytes (Tregs) is responsible for regulating the inflammatory response and maintaining homeostasis of microbiota and is also dependent on intestinal microbiome composition during the early period of life [318][319]. Bacterial metabolites are also involved in regulation of the effect of the immune system on intestinal mucosa. Short-chain fatty acids decrease abundance of Th1/Th17 T helpers while increasing the presence of Tregs, thus reducing the intensity of inflammatory response [320][321]. Secondary bile acids were also able to modulate differentiation of Th17 and RORγ+ Tregs [322][323]. The relative abundance of different components of the immune system plays a crucial role in the determination of the outcome of immune response: an increase in abundance of tumor-associated macrophages and fibroblasts, Tregs, regulatory B cells, and myeloid-derived suppressor cells (MDSCs) is associated with rapid and unimpeded tumor growth [324], while increase in the abundance of CD8+ cytotoxic T lymphocytes, natural killer (NK) cells, and M1 macrophages and CD4+ T lymphocytes is associated with the direct destruction of tumor cells or with stimulation of this process [325][326].
Plant polyphenolic compounds can modulate tumor microenvironments either indirectly, by regulating the composition of intestinal microbiota, or directly, by mediating interactions between different types of immune cells. For example, polyphenols can stabilize the relative abundance of different immune cells on the intestinal mucosa and prevent development of chronic inflammation by preventing carcinogen-induced dysbiosis. Moreover, maintenance of intestinal eubiosis can be associated with sensitivity to immune checkpoint therapy; however, the exact molecular mechanisms responsible for this association are yet to be determined [327].
Mechanisms of the modulation of tumor immune microenvironments by plant phenolic compounds are not limited to prevention of dysbiosis. During multiple in vitro experiments, an increase in the abundance of CD8+ T lymphocytes and enhancement of their cytotoxic activity upon polyphenol treatment were observed [328]. Thus, treatment of the murine Lewis lung adenocarcinoma model with curcumin led to suppression of MDSCs, resulting in an increase in the number of CD8+ T lymphocytes [329]. Cytotoxic activity of NK cells can also be increased by natural phenolic compounds: treatment of human L3.4 pancreatic ductal adenocarcinoma cell culture and MIA PaCa-2 pancreatic adenocarcinoma cell culture with curcuminoids led to the enhancement of cytotoxic activity of co-cultured donor NK cells [330]. Immunomodulatory effects of plant phenolic compounds that increase the anti-tumor immune response were also observed during in vivo studies. Treatment of mice xenografted with B16F10 murine melanoma cells with resveratrol led to the increase in intra-tumoral NK cell cytotoxicity; stimulation with IL-2 additionally enhanced this effect [331]. Life-long intake of genistein by rats resulted in lower incidence of mammary tumors upon stimulation of tumorigenesis by dimethylbenzanthracene, as well as the suppression of CD4+ Tregs proliferation and an increase in the abundance of CD8+ cytotoxic T lymphocytes, thus resulting in enhancement of the sensitivity of these tumors to tamoxifen [332].
Interactions between immune cells in tumor microenvironments are targeted by the immune checkpoint inhibitors—anticancer drugs that prevent the suppression of cytotoxic T lymphocyte activity [333]. These drugs can be used in combination with other types of anticancer therapy, such as photodynamic therapy [334]. Among several different immune checkpoints, the system that consists of PD-1 receptor and PD-L1 ligand system is the most studied to date. Activation of membrane PD-1 of T lymphocytes leads to a decrease in their activity, proliferation, and survival [335]. Recent studies indicate that plant phenolic compounds can modulate this regulatory mechanism. Thus, resveratrol was able to attenuate its activity by preventing PD-L1 glycosylation and dimerization in JIMT-1 human breast adenocarcinoma cells that overexpress this ligand [336]. Treatment with apigenin prevented induction of PD-L1 by γ-interferon in several melanoma cell cultures in vitro, and enhanced infiltration of T lymphocytes in murine xenografts of these melanoma cells, while simultaneously suppressing expression of PD-L1 in dendritic cells [337]. The similar activity of apigenin, along with luteolin, towards KRAS-mutated non-small cell lung carcinoma cell cultures and xenografts was described: these compounds were able to prevent γ-interferon-induced upregulation of PD-L1 expression by suppressing activity of the MUC-1C/STAT3 signaling pathway; molecular docking results suggest that these polyphenols can interact directly with STAT3 molecules [338]. Curcumin was also able to downregulate PD-L1 expression in Cal27 and FaDu oral squamous cell carcinoma cultures, as well as in the murine model of induction of oral squamous cell carcinoma by 4-nitroquinoline oxide [339]. This phenolic compound also demonstrated similar PD-L1 downregulation in Hep3B and CSQT-2 hepatocellular carcinoma cell cultures, an exerted synergistic effect with the anti-PD-1 therapy of Hep3B murine xenografts [340]. Several other polyphenolic compounds have inhibitory activity towards the PD-1/PD-L1 immune checkpoint; EGCG, hesperidin, baicalin, and quercitin are the examples of these polyphenols [341]. At the same time, it is important to emphasize that plant phenolic compounds can also demonstrate the opposite activity: thus, treatment of A549, H460, and H1299 lung adenocarcinoma cell cultures with resveratrol led to an increase in Snail protein stability, which in turn led to the stimulation of the Wnt signaling pathway, and ultimately, upregulated the PD-L1 expression [342]. These data point out the necessity of further investigation of PD-1/PD-L1 immune checkpoint functioning, and of the mechanisms of plant polyphenol-mediated changes in this regulatory system.
When considering the antitumor and chemopreventive activity of plant polyphenols, another important aspect should be taken into account: natural phenolic compounds are not metabolically inert, and may undergo extensive chemical modification by host microbiota [343]. It is established that the products of the bacterial metabolism of polyphenols might have different activities as compared to the original compounds [344]. The most prominent examples of such metabolites are equol [345][346] and urolithins [347][348], products of the modification of daidzein and ellagitannins, respectively. Their antitumor and chemopreventive activity was demonstrated in a number of studies.
Treatment of MCF-7 human beast adenocarcinoma cell culture resulted in the suppression of the proliferation and induction of apoptosis by upregulating miR-10a-5p expression and subsequent inhibition of the PI3K/AKT signaling pathway [349]. In HCT-15 colon cancer cell cultures, equol was able to enhance expression of Nrf2, thus suppressing proliferation of this culture [350]. Equol also exerted antimetastatic activity: treatment of MDA-MB-231 human breast cancer cell culture with this compound led to a decrease in the invasiveness of these cells by inhibiting MMP-2 expression [351]. Treatment of MCF10 human breast cancer cell culture with equol led to abrogation of TNF-1-mediated activation of Gli1, a member of the hedgehog signaling pathway, which is responsible for induction of cellular motility [352]. The chemopreventive activity of equol was also described: dietary supplementation of rats with this compound alleviated the efficiency of dimethylbenzanthracene-induced carcinogenesis [353]. Similar dietary supplementation reduced the efficiency of carcinogenesis by urethane in mice, while simultaneously increasing the level of superoxide dismutase and reducing the concentration of oxidative stress markers in blood serum [354].
The anticancer activity of urolithins was also demonstrated: treatment of Caco-2 colon cancer cell culture with these compounds resulted in suppression of the proliferation and induction of apoptosis [355], likely caused by the upregulation of CDKN1A expression [356]. In 22RV1 and LNCaP human prostate cancer cell cultures, various urolithins were able to induce apoptosis and to increase p53 expression [357]. The similar activity of these substances towards the expression of p53 in colon cancer cell cultures was described [358]. Treatment of these cultures with urolithin also led to cell cycle arrest, apoptosis induction, and an increase in ROS production. Interestingly, treatment of normal human fibroblast cell cultures with this phenolic compound had the opposite effect on ROS production [359]. Dependence of the direction of oxidative stress modulation by urolithins on the type of cell culture was demonstrated in a number of studies, and the mechanisms behind this phenomenon include the condition of cellular antioxidants and oxidative stress-inducing factors [360]. The proapoptotic and antiproliferative activity of urolithins is not limited to the mechanisms mentioned above: these substances might also modulate PI3K/AKT, NF-κB, and WNT/β-catenin signaling pathways [348].
References
- Breasted, J.H. The Edwin Smith Surgical Papyrus: Published in Facsimile and Hieroglyphic Transliteration with Translation and Commentary in Two Volumes; University of Chicago Oriental Institute Publications; University of Chicago Press: Chicago, IL, USA, 1991; ISBN 978-0-918986-73-3.
- Stratton, M.R. Exploring the Genomes of Cancer Cells: Progress and Promise. Science 2011, 331, 1553–1558.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558.
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898.
- Loeb, L.A.; Harris, C.C. Advances in Chemical Carcinogenesis: A Historical Review and Prospective. Cancer Res. 2008, 68, 6863–6872.
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292.
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of Rare Circulating Tumour Cells in Cancer Patients by Microchip Technology. Nature 2007, 450, 1235–1239.
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only Three Driver Gene Mutations Are Required for the Development of Lung and Colorectal Cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123.
- Hanahan, D. Rethinking the War on Cancer. Lancet 2014, 383, 558–563.
- Block, K.I.; Gyllenhaal, C.; Lowe, L.; Amedei, A.; Amin, A.R.M.R.; Amin, A.; Aquilano, K.; Arbiser, J.; Arreola, A.; Arzumanyan, A.; et al. Designing a Broad-Spectrum Integrative Approach for Cancer Prevention and Treatment. Semin. Cancer Biol. 2015, 35, S276–S304.
- Song, M.; Vogelstein, B.; Giovannucci, E.L.; Willett, W.C.; Tomasetti, C. Cancer Prevention: Molecular and Epidemiologic Consensus. Science 2018, 361, 1317–1318.
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334.
- Kirtonia, A.; Sethi, G.; Garg, M. The Multifaceted Role of Reactive Oxygen Species in Tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483.
- Chen, C.; Kong, A.-N.T. Dietary Cancer-Chemopreventive Compounds: From Signaling and Gene Expression to Pharmacological Effects. Trends Pharm. Sci. 2005, 26, 318–326.
- Rather, R.A.; Bhagat, M. Cancer Chemoprevention and Piperine: Molecular Mechanisms and Therapeutic Opportunities. Front. Cell. Dev. Biol. 2018, 6, 10.
- Workman, P.; Draetta, G.F.; Schellens, J.H.M.; Bernards, R. How Much Longer Will We Put Up with $100,000 Cancer Drugs? Cell 2017, 168, 579–583.
- Erb, M.; Kliebenstein, D.J. Plant Secondary Metabolites as Defenses, Regulators, and Primary Metabolites: The Blurred Functional Trichotomy. Plant Physiol. 2020, 184, 39–52.
- Valdés-Jiménez, A.; Peña-Varas, C.; Borrego-Muñoz, P.; Arrue, L.; Alegría-Arcos, M.; Nour-Eldin, H.; Dreyer, I.; Nuñez-Vivanco, G.; Ramírez, D. PSC-Db: A Structured and Searchable 3D-Database for Plant Secondary Compounds. Molecules 2021, 26, 1124.
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of Plant Defense against Insect Herbivores. Plant Signal. Behav. 2012, 7, 1306–1320.
- Cheynier, V.; Comte, G.; Davies, K.M.; Lattanzio, V.; Martens, S. Plant Phenolics: Recent Advances on Their Biosynthesis, Genetics, and Ecophysiology. Plant Physiol. Biochem. 2013, 72, 1–20.
- El Gharras, H. Polyphenols: Food Sources, Properties and Applications-a Review: Nutraceutical Polyphenols. Int. J. Food Sci. Technol. 2009, 44, 2512–2518.
- De la Rosa, L.A.; Alvarez-Parrilla, E.; Gonzlez-Aguilar, G.A. (Eds.) Fruit and Vegetable Phytochemicals; Wiley-Blackwell: Oxford, UK, 2009; ISBN 978-0-8138-0939-7.
- Khoddami, A.; Wilkes, M.; Roberts, T. Techniques for Analysis of Plant Phenolic Compounds. Molecules 2013, 18, 2328–2375.
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures. J. AOAC Int. 2019, 102, 1397–1400.
- Soto-Vaca, A.; Gutierrez, A.; Losso, J.N.; Xu, Z.; Finley, J.W. Evolution of Phenolic Compounds from Color and Flavor Problems to Health Benefits. J. Agric. Food Chem. 2012, 60, 6658–6677.
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food Sources and Bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747.
- Corcoran, M.P.; McKay, D.L.; Blumberg, J.B. Flavonoid Basics: Chemistry, Sources, Mechanisms of Action, and Safety. J. Nutr. Gerontol. Geriatr. 2012, 31, 176–189.
- Pérez-Jiménez, J.; Neveu, V.; Vos, F.; Scalbert, A. Identification of the 100 Richest Dietary Sources of Polyphenols: An Application of the Phenol-Explorer Database. Eur. J. Clin. Nutr. 2010, 64, S112–S120.
- Lambert, J.D.; Lee, M.-J.; Lu, H.; Meng, X.; Hong, J.J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-Gallate Is Absorbed but Extensively Glucuronidated Following Oral Administration to Mice. J. Nutr. 2003, 133, 4172–4177.
- Rotches-Ribalta, M.; Andres-Lacueva, C.; Estruch, R.; Escribano, E.; Urpi-Sarda, M. Pharmacokinetics of Resveratrol Metabolic Profile in Healthy Humans after Moderate Consumption of Red Wine and Grape Extract Tablets. Pharmacol. Res. 2012, 66, 375–382.
- Servili, M.; Sordini, B.; Esposto, S.; Urbani, S.; Veneziani, G.; Di Maio, I.; Selvaggini, R.; Taticchi, A. Biological Activities of Phenolic Compounds of Extra Virgin Olive Oil. Antioxidants 2013, 3, 1–23.
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-From Molecule to Biological Function. Angew. Chem. Int. Ed. 2012, 51, 5308–5332.
- Van Wyk, B.-E.; Wink, M. Medicinal Plants of the World: An Illustrated Scientific Guide to Important Medicinal Plants and Their Uses, 1st ed.; Timber Press: Portland, OR, USA, 2004; ISBN 978-0-88192-602-6.
- Kim, S.H.; Kim, M.S.; Lee, M.S.; Park, Y.S.; Lee, H.J.; Kang, S.; Lee, H.S.; Lee, K.-E.; Yang, H.J.; Kim, M.J.; et al. Korean Diet: Characteristics and Historical Background. J. Ethn. Foods 2016, 3, 26–31.
- Pallauf, K.; Giller, K.; Huebbe, P.; Rimbach, G. Nutrition and Healthy Ageing: Calorie Restriction or Polyphenol-Rich “MediterrAsian” Diet? Oxidative Med. Cell. Longev. 2013, 2013, 707421.
- Amiot, M.J.; Riva, C.; Vinet, A. Effects of Dietary Polyphenols on Metabolic Syndrome Features in Humans: A Systematic Review. Obes. Rev. 2016, 17, 573–586.
- Rienks, J.; Barbaresko, J.; Oluwagbemigun, K.; Schmid, M.; Nöthlings, U. Polyphenol Exposure and Risk of Type 2 Diabetes: Dose-Response Meta-Analyses and Systematic Review of Prospective Cohort Studies. Am. J. Clin. Nutr. 2018, 108, 49–61.
- Stefani, M.; Rigacci, S. Beneficial Properties of Natural Phenols: Highlight on Protection against Pathological Conditions Associated with Amyloid Aggregation. Biofactors 2014, 40, 482–493.
- Feinleib, M. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. JAMA 1981, 245, 511.
- Grosso, G.; Godos, J.; Lamuela-Raventos, R.; Ray, S.; Micek, A.; Pajak, A.; Sciacca, S.; D’Orazio, N.; Del Rio, D.; Galvano, F. A Comprehensive Meta-Analysis on Dietary Flavonoid and Lignan Intake and Cancer Risk: Level of Evidence and Limitations. Mol. Nutr. Food Res. 2017, 61, 1600930.
- Patra, S.; Pradhan, B.; Nayak, R.; Behera, C.; Das, S.; Patra, S.K.; Efferth, T.; Jena, M.; Bhutia, S.K. Dietary Polyphenols in Chemoprevention and Synergistic Effect in Cancer: Clinical Evidences and Molecular Mechanisms of Action. Phytomedicine 2021, 90, 153554.
- Kirsanov, K.I.; Vlasova, O.A.; Fetisov, T.I.; Zenkov, R.G.; Lesovaya, E.A.; Belitsky, G.A.; Gurova, K.; Yakubovskaya, M.G. Influence of DNA-Binding Compounds with Cancer Preventive Activity on the Mechanisms of Gene Expression Regulation. Adv. Mol. Onkol. 2019, 5, 41–63.
- Hazafa, A.; Rehman, K.-U.; Jahan, N.; Jabeen, Z. The Role of Polyphenol (Flavonoids) Compounds in the Treatment of Cancer Cells. Nutr. Cancer 2020, 72, 386–397.
- Chou, C.-C.; Yang, J.-S.; Lu, H.-F.; Ip, S.-W.; Lo, C.; Wu, C.-C.; Lin, J.-P.; Tang, N.-Y.; Chung, J.-G.; Chou, M.-J.; et al. Quercetin-Mediated Cell Cycle Arrest and Apoptosis Involving Activation of a Caspase Cascade through the Mitochondrial Pathway in Human Breast Cancer MCF-7 Cells. Arch. Pharm. Res. 2010, 33, 1181–1191.
- Duo, J.; Ying, G.-G.; Wang, G.-W.; Zhang, L. Quercetin Inhibits Human Breast Cancer Cell Proliferation and Induces Apoptosis via Bcl-2 and Bax Regulation. Mol. Med. Rep. 2012, 5, 1453–1456.
- Adhami, V.M.; Malik, A.; Zaman, N.; Sarfaraz, S.; Siddiqui, I.A.; Syed, D.N.; Afaq, F.; Pasha, F.S.; Saleem, M.; Mukhtar, H. Combined Inhibitory Effects of Green Tea Polyphenols and Selective Cyclooxygenase-2 Inhibitors on the Growth of Human Prostate Cancer Cells Both in Vitro and in Vivo. Clin. Cancer Res. 2007, 13, 1611–1619.
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Mechanism of Transforming Growth Factor-Beta1-Induced Expression of Vascular Endothelial Growth Factor in Murine Osteoblastic MC3T3-E1 Cells. Biochim. Biophys. Acta 2000, 1497, 69–76.
- Chadalapaka, G.; Jutooru, I.; Chintharlapalli, S.; Papineni, S.; Smith, R.; Li, X.; Safe, S. Curcumin Decreases Specificity Protein Expression in Bladder Cancer Cells. Cancer Res. 2008, 68, 5345–5354.
- Namasivayam, N. Chemoprevention in Experimental Animals: Chemoprevention in Experimental Animals. Ann. N. Y. Acad. Sci. 2011, 1215, 60–71.
- Zhou, Y.; Zheng, J.; Li, Y.; Xu, D.-P.; Li, S.; Chen, Y.-M.; Li, H.-B. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients 2016, 8, 515.
- Kuroiwa, Y.; Nishikawa, A.; Kitamura, Y.; Kanki, K.; Ishii, Y.; Umemura, T.; Hirose, M. Protective Effects of Benzyl Isothiocyanate and Sulforaphane but Not Resveratrol against Initiation of Pancreatic Carcinogenesis in Hamsters. Cancer Lett. 2006, 241, 275–280.
- Sengottuvelan, M.; Senthilkumar, R.; Nalini, N. Modulatory Influence of Dietary Resveratrol during Different Phases of 1,2-Dimethylhydrazine Induced Mucosal Lipid-Peroxidation, Antioxidant Status and Aberrant Crypt Foci Development in Rat Colon Carcinogenesis. Biochim. Biophys. Acta 2006, 1760, 1175–1183.
- Mazué, F.; Delmas, D.; Murillo, G.; Saleiro, D.; Limagne, E.; Latruffe, N. Differential Protective Effects of Red Wine Polyphenol Extracts (RWEs) on Colon Carcinogenesis. Food Funct. 2014, 5, 663–670.
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein Decreases the Breast Cancer Stem-like Cell Population through Hedgehog Pathway. Stem Cell. Res. Ther. 2013, 4, 146.
- Kim, H.-S.; Wannatung, T.; Lee, S.; Yang, W.K.; Chung, S.H.; Lim, J.-S.; Choe, W.; Kang, I.; Kim, S.-S.; Ha, J. Quercetin Enhances Hypoxia-Mediated Apoptosis via Direct Inhibition of AMPK Activity in HCT116 Colon Cancer. Apoptosis 2012, 17, 938–949.
- Thomasset, S.C.; Berry, D.P.; Garcea, G.; Marczylo, T.; Steward, W.P.; Gescher, A.J. Dietary Polyphenolic Phytochemicals--Promising Cancer Chemopreventive Agents in Humans? A Review of Their Clinical Properties. Int. J. Cancer 2007, 120, 451–458.
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharm. 2019, 10, 1614.
- Lazarevic, B.; Boezelijn, G.; Diep, L.M.; Kvernrod, K.; Ogren, O.; Ramberg, H.; Moen, A.; Wessel, N.; Berg, R.E.; Egge-Jacobsen, W.; et al. Efficacy and Safety of Short-Term Genistein Intervention in Patients with Localized Prostate Cancer Prior to Radical Prostatectomy: A Randomized, Placebo-Controlled, Double-Blind Phase 2 Clinical Trial. Nutr. Cancer 2011, 63, 889–898.
- Carroll, R.E.; Benya, R.V.; Turgeon, D.K.; Vareed, S.; Neuman, M.; Rodriguez, L.; Kakarala, M.; Carpenter, P.M.; McLaren, C.; Meyskens, F.L.; et al. Phase IIa Clinical Trial of Curcumin for the Prevention of Colorectal Neoplasia. Cancer Prev. Res. 2011, 4, 354–364.
- Kiselev, V.I.; Ashrafyan, L.A.; Muyzhnek, E.L.; Gerfanova, E.V.; Antonova, I.B.; Aleshikova, O.I.; Sarkar, F.H. A New Promising Way of Maintenance Therapy in Advanced Ovarian Cancer: A Comparative Clinical Study. BMC Cancer 2018, 18, 904.
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.-A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I Dose Escalation Trial of Docetaxel plus Curcumin in Patients with Advanced and Metastatic Breast Cancer. Cancer Biol. Ther. 2010, 9, 8–14.
- Trudel, D.; Labbé, D.P.; Araya-Farias, M.; Doyen, A.; Bazinet, L.; Duchesne, T.; Plante, M.; Grégoire, J.; Renaud, M.-C.; Bachvarov, D.; et al. A Two-Stage, Single-Arm, Phase II Study of EGCG-Enriched Green Tea Drink as a Maintenance Therapy in Women with Advanced Stage Ovarian Cancer. Gynecol. Oncol. 2013, 131, 357–361.
- Nguyen, M.M.; Ahmann, F.R.; Nagle, R.B.; Hsu, C.-H.; Tangrea, J.A.; Parnes, H.L.; Sokoloff, M.H.; Gretzer, M.B.; Chow, H.-H.S. Randomized, Double-Blind, Placebo-Controlled Trial of Polyphenon E in Prostate Cancer Patients before Prostatectomy: Evaluation of Potential Chemopreventive Activities. Cancer Prev. Res. 2012, 5, 290–298.
- Gontero, P.; Marra, G.; Soria, F.; Oderda, M.; Zitella, A.; Baratta, F.; Chiorino, G.; Gregnanin, I.; Daniele, L.; Cattel, L.; et al. A Randomized Double-Blind Placebo Controlled Phase I-II Study on Clinical and Molecular Effects of Dietary Supplements in Men with Precancerous Prostatic Lesions. Chemoprevention or “Chemopromotion”? Prostate 2015, 75, 1177–1186.
- Wink, M. Modes of Action of Herbal Medicines and Plant Secondary Metabolites. Medicines 2015, 2, 251–286.
- Sah, J.F.; Balasubramanian, S.; Eckert, R.L.; Rorke, E.A. Epigallocatechin-3-Gallate Inhibits Epidermal Growth Factor Receptor Signaling Pathway. J. Biol. Chem. 2004, 279, 12755–12762.
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester Bond-Containing Tea Polyphenols Potently Inhibit Proteasome Activity in Vitro and in Vivo. J. Biol. Chem. 2001, 276, 13322–13330.
- Leone, M.; Zhai, D.; Sareth, S.; Kitada, S.; Reed, J.C.; Pellecchia, M. Cancer Prevention by Tea Polyphenols Is Linked to Their Direct Inhibition of Antiapoptotic Bcl-2-Family Proteins. Cancer Res. 2003, 63, 8118–8121.
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A Receptor for Green Tea Polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381.
- Shimizu, M.; Deguchi, A.; Hara, Y.; Moriwaki, H.; Weinstein, I.B. EGCG Inhibits Activation of the Insulin-like Growth Factor-1 Receptor in Human Colon Cancer Cells. Biochem. Biophys. Res. Commun. 2005, 334, 947–953.
- Kuzuhara, T.; Suganuma, M.; Fujiki, H. Green Tea Catechin as a Chemical Chaperone in Cancer Prevention. Cancer Lett. 2008, 261, 12–20.
- Fujimura, Y.; Tachibana, H.; Yamada, K. Lipid Raft-Associated Catechin Suppresses the FcϵRI Expression by Inhibiting Phosphorylation of the Extracellular Signal-Regulated Kinase1/2. FEBS Lett. 2004, 556, 204–210.
- Tu, B.; Liu, Z.-J.; Chen, Z.-F.; Ouyang, Y.; Hu, Y.-J. Understanding the Structure–Activity Relationship between Quercetin and Naringenin: In Vitro. RSC Adv. 2015, 5, 106171–106181.
- Nair, M.S.; Shukla, A. Molecular Modeling, Simulation and Principal Component Analysis of Binding of Resveratrol and Its Analogues with DNA. J. Biomol. Struct. Dyn. 2020, 38, 3087–3097.
- N’soukpoé-Kossi, C.N.; Bourassa, P.; Mandeville, J.S.; Bekale, L.; Bariyanga, J.; Tajmir-Riahi, H.A. Locating the Binding Sites of Antioxidants Resveratrol, Genistein and Curcumin with TRNA. Int. J. Biol. Macromol. 2015, 80, 41–47.
- Platella, C.; Raucci, U.; Rega, N.; D’Atri, S.; Levati, L.; Roviello, G.N.; Fuggetta, M.P.; Musumeci, D.; Montesarchio, D. Shedding Light on the Interaction of Polydatin and Resveratrol with G-Quadruplex and Duplex DNA: A Biophysical, Computational and Biological Approach. Int. J. Biol. Macromol. 2020, 151, 1163–1172.
- Luch, A.; Baird, W.M. Carcinogenic Polycyclic Aromatic Hydrocarbons. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 85–123. ISBN 978-0-08-046884-6.
- Gibis, M. Heterocyclic Aromatic Amines in Cooked Meat Products: Causes, Formation, Occurrence, and Risk Assessment: Heterocyclic Amines in Cooked Meat Products…. Compr. Rev. Food Sci. Food Saf. 2016, 15, 269–302.
- Attaluri, S.; Bonala, R.R.; Yang, I.-Y.; Lukin, M.A.; Wen, Y.; Grollman, A.P.; Moriya, M.; Iden, C.R.; Johnson, F. DNA Adducts of Aristolochic Acid II: Total Synthesis and Site-Specific Mutagenesis Studies in Mammalian Cells. Nucleic Acids Res. 2010, 38, 339–352.
- Wu, H.-C.; Santella, R. The Role of Aflatoxins in Hepatocellular Carcinoma. Hepat. Mon. 2012, 12.
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities. Toxicol. Sci. 2011, 120, S49–S75.
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The Aryl Hydrocarbon Receptor Complex and the Control of Gene Expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250.
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the Same but Different: Promiscuity and Diversity in the Molecular Mechanisms of Action of the Aryl Hydrocarbon (Dioxin) Receptor. Toxicol. Sci. 2011, 124, 1–22.
- Lindsey, S.; Papoutsakis, E.T. The Evolving Role of the Aryl Hydrocarbon Receptor (AHR) in the Normophysiology of Hematopoiesis. Stem Cell. Rev. Rep. 2012, 8, 1223–1235.
- Xue, Z.; Li, D.; Yu, W.; Zhang, Q.; Hou, X.; He, Y.; Kou, X. Mechanisms and Therapeutic Prospects of Polyphenols as Modulators of the Aryl Hydrocarbon Receptor. Food Funct. 2017, 8, 1414–1437.
- Ciolino, H.P.; Daschner, P.J.; Wang, T.T.Y.; Yeh, G.C. Effect of Curcumin on the Aryl Hydrocarbon Receptor and Cytochrome P450 1A1 in MCF-7 Human Breast Carcinoma Cells. Biochem. Pharmacol. 1998, 56, 197–206.
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Dietary Flavonols Quercetin and Kaempferol Are Ligands of the Aryl Hydrocarbon Receptor That Affect CYP1A1 Transcription Differentially. Biochem. J. 1999, 340 Pt 3, 715–722.
- Perdew, G.H.; Hollingshead, B.D.; Dinatale, B.C.; Morales, J.L.; Labrecque, M.P.; Takhar, M.K.; Tam, K.J.; Beischlag, T.V. Estrogen Receptor Expression Is Required for Low-Dose Resveratrol-Mediated Repression of Aryl Hydrocarbon Receptor Activity. J. Pharm. Exp. Ther. 2010, 335, 273–283.
- Fukuda, I.; Mukai, R.; Kawase, M.; Yoshida, K.; Ashida, H. Interaction between the Aryl Hydrocarbon Receptor and Its Antagonists, Flavonoids. Biochem. Biophys. Res. Commun. 2007, 359, 822–827.
- Jin, U.-H.; Park, H.; Li, X.; Davidson, L.A.; Allred, C.; Patil, B.; Jayaprakasha, G.; Orr, A.A.; Mao, L.; Chapkin, R.S.; et al. Structure-Dependent Modulation of Aryl Hydrocarbon Receptor-Mediated Activities by Flavonoids. Toxicol. Sci. 2018, 164, 205–217.
- Kaur, M.; Badhan, R.K.S. Phytochemical Mediated-Modulation of the Expression and Transporter Function of Breast Cancer Resistance Protein at the Blood-Brain Barrier: An in-Vitro Study. Brain Res. 2017, 1654, 9–23.
- Goya-Jorge, E.; Giner, R.M.; Sylla-Iyarreta Veitía, M.; Gozalbes, R.; Barigye, S.J. Predictive Modeling of Aryl Hydrocarbon Receptor (AhR) Agonism. Chemosphere 2020, 256, 127068.
- Goya-Jorge, E.; Jorge Rodríguez, M.E.; Veitía, M.S.-I.; Giner, R.M. Plant Occurring Flavonoids as Modulators of the Aryl Hydrocarbon Receptor. Molecules 2021, 26, 2315.
- Mukai, R.; Shirai, Y.; Saito, N.; Fukuda, I.; Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Suppression Mechanisms of Flavonoids on Aryl Hydrocarbon Receptor-Mediated Signal Transduction. Arch. Biochem. Biophys. 2010, 501, 134–141.
- Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Curcumin Suppresses the Transformation of an Aryl Hydrocarbon Receptor through Its Phosphorylation. Arch. Biochem. Biophys. 2007, 466, 267–273.
- Quadri, S.A.; Qadri, A.N.; Hahn, M.E.; Mann, K.K.; Sherr, D.H. The Bioflavonoid Galangin Blocks Aryl Hydrocarbon Receptor Activation and Polycyclic Aromatic Hydrocarbon-Induced Pre-B Cell Apoptosis. Mol. Pharm. 2000, 58, 515–525.
- Palermo, C.M.; Westlake, C.A.; Gasiewicz, T.A. Epigallocatechin Gallate Inhibits Aryl Hydrocarbon Receptor Gene Transcription through an Indirect Mechanism Involving Binding to a 90 KDa Heat Shock Protein. Biochemistry 2005, 44, 5041–5052.
- Ciolino, H.P.; Daschner, P.J.; Yeh, G.C. Resveratrol Inhibits Transcription of CYP1A1 in Vitro by Preventing Activation of the Aryl Hydrocarbon Receptor. Cancer Res. 1998, 58, 5707–5712.
- Froyen, E.B.; Steinberg, F.M. Genistein Decreases Basal Hepatic Cytochrome P450 1A1 Protein Expression and Activity in Swiss Webster Mice. Nutr. Res. 2016, 36, 430–439.
- Macpherson, L.; Matthews, J. Inhibition of Aryl Hydrocarbon Receptor-Dependent Transcription by Resveratrol or Kaempferol Is Independent of Estrogen Receptor α Expression in Human Breast Cancer Cells. Cancer Lett. 2010, 299, 119–129.
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54.
- Wahlang, B.; Falkner, K.C.; Cave, M.C.; Prough, R.A. Role of Cytochrome P450 Monooxygenase in Carcinogen and Chemotherapeutic Drug Metabolism. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 74, pp. 1–33. ISBN 978-0-12-803119-3.
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure−Function Relationships of Inhibition of Human Cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 Flavonoid Derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935.
- Kimura, Y.; Ito, H.; Ohnishi, R.; Hatano, T. Inhibitory Effects of Polyphenols on Human Cytochrome P450 3A4 and 2C9 Activity. Food Chem. Toxicol. 2010, 48, 429–435.
- Matsuno, Y.; Atsumi, Y.; Alauddin, M.; Rana, M.M.; Fujimori, H.; Hyodo, M.; Shimizu, A.; Ikuta, T.; Tani, H.; Torigoe, H.; et al. Resveratrol and Its Related Polyphenols Contribute to the Maintenance of Genome Stability. Sci. Rep. 2020, 10, 5388.
- Yan, H.; Jiang, J.; Du, A.; Gao, J.; Zhang, D.; Song, L. Genistein Enhances Radiosensitivity of Human Hepatocellular Carcinoma Cells by Inducing G2/M Arrest and Apoptosis. Radiat. Res. 2020, 193, 286.
- Majidinia, M.; Bishayee, A.; Yousefi, B. Polyphenols: Major Regulators of Key Components of DNA Damage Response in Cancer. DNA Repair. 2019, 82, 102679.
- Zhou, Z.; Liu, Y.; Jiang, X.; Zheng, C.; Luo, W.; Xiang, X.; Qi, X.; Shen, J. Metformin Modified Chitosan as a Multi-Functional Adjuvant to Enhance Cisplatin-Based Tumor Chemotherapy Efficacy. Int. J. Biol. Macromol. 2023, 224, 797–809.
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of Oxidative Stress and DNA Damage in Human Carcinogenesis. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 193–201.
- Gates, K.S. An Overview of Chemical Processes That Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reactions with Radicals. Chem. Res. Toxicol. 2009, 22, 1747–1760.
- Pereira, C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative Stress and DNA Damage: Implications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417.
- Jomova, K.; Valko, M. Advances in Metal-Induced Oxidative Stress and Human Disease. Toxicology 2011, 283, 65–87.
- Gebicki, J.M. Oxidative Stress, Free Radicals and Protein Peroxides. Arch. Biochem. Biophys. 2016, 595, 33–39.
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679.
- Cos, P.; Ying, L.; Calomme, M.; Hu, J.P.; Cimanga, K.; Van Poel, B.; Pieters, L.; Vlietinck, A.J.; Vanden Berghe, D. Structure-Activity Relationship and Classification of Flavonoids as Inhibitors of Xanthine Oxidase and Superoxide Scavengers. J. Nat. Prod. 1998, 61, 71–76.
- Chen, J.-W.; Zhu, Z.-Q.; Hu, T.-X.; Zhu, D.-Y. Structure-Activity Relationship of Natural Flavonoids in Hydroxyl Radical-Scavenging Effects. Acta Pharm. Sin. 2002, 23, 667–672.
- Olszowy, M. What Is Responsible for Antioxidant Properties of Polyphenolic Compounds from Plants? Plant Physiol. Biochem. 2019, 144, 135–143.
- Guo, Q.; Zhao, B.; Shen, S.; Hou, J.; Hu, J.; Xin, W. ESR Study on the Structure–Antioxidant Activity Relationship of Tea Catechins and Their Epimers. Biochim. Biophys. Acta (BBA) Gen. Subj. 1999, 1427, 13–23.
- Nakagawa, T.; Yokozawa, T. Direct Scavenging of Nitric Oxide and Superoxide by Green Tea. Food Chem. Toxicol. 2002, 40, 1745–1750.
- Shanmugam, T.; Selvaraj, M.; Poomalai, S. Epigallocatechin Gallate Potentially Abrogates Fluoride Induced Lung Oxidative Stress, Inflammation via Nrf2/Keap1 Signaling Pathway in Rats: An in-Vivo and in-Silico Study. Int. Immunopharmacol. 2016, 39, 128–139.
- Giftson, J.S.; Jayanthi, S.; Nalini, N. Chemopreventive Efficacy of Gallic Acid, an Antioxidant and Anticarcinogenic Polyphenol, against 1,2-Dimethyl Hydrazine Induced Rat Colon Carcinogenesis. Investig. New Drugs 2010, 28, 251–259.
- Sharmila, G.; Athirai, T.; Kiruthiga, B.; Senthilkumar, K.; Elumalai, P.; Arunkumar, R.; Arunakaran, J. Chemopreventive Effect of Quercetin in MNU and Testosterone Induced Prostate Cancer of Sprague-Dawley Rats. Nutr. Cancer 2014, 66, 38–46.
- Henning, S.M.; Niu, Y.; Lee, N.H.; Thames, G.D.; Minutti, R.R.; Wang, H.; Go, V.L.W.; Heber, D. Bioavailability and Antioxidant Activity of Tea Flavanols after Consumption of Green Tea, Black Tea, or a Green Tea Extract Supplement. Am. J. Clin. Nutr. 2004, 80, 1558–1564.
- Young, J.F.; Dragsted, L.O.; Haraldsdóttir, J.; Daneshvar, B.; Kall, M.A.; Loft, S.; Nilsson, L.; Nielsen, S.E.; Mayer, B.; Skibsted, L.H.; et al. Green Tea Extract Only Affects Markers of Oxidative Status Postprandially: Lasting Antioxidant Effect of Flavonoid-Free Diet. Br. J. Nutr. 2002, 87, 343–355.
- Fallah, A.A.; Sarmast, E.; Jafari, T. Effect of Dietary Anthocyanins on Biomarkers of Oxidative Stress and Antioxidative Capacity: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Funct. Foods 2020, 68, 103912.
- Mira, L.; Tereza Fernandez, M.; Santos, M.; Rocha, R.; Helena Florêncio, M.; Jennings, K.R. Interactions of Flavonoids with Iron and Copper Ions: A Mechanism for Their Antioxidant Activity. Free Radic. Res. 2002, 36, 1199–1208.
- Adjimani, J.P.; Asare, P. Antioxidant and Free Radical Scavenging Activity of Iron Chelators. Toxicol. Rep. 2015, 2, 721–728.
- McCubrey, J.A.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Yang, L.V.; Murata, R.M.; Rosalen, P.L.; Scalisi, A.; Neri, L.M.; Cocco, L.; et al. Effects of Resveratrol, Curcumin, Berberine and Other Nutraceuticals on Aging, Cancer Development, Cancer Stem Cells and MicroRNAs. Aging 2017, 9, 1477–1536.
- León-González, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-Oxidant Activity of Polyphenols and Its Implication on Cancer Chemoprevention and Chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380.
- Kim, H.-S.; Quon, M.J.; Kim, J. New Insights into the Mechanisms of Polyphenols beyond Antioxidant Properties; Lessons from the Green Tea Polyphenol, Epigallocatechin 3-Gallate. Redox Biol. 2014, 2, 187–195.
- He, F.; Antonucci, L.; Karin, M. NRF2 as a Regulator of Cell Metabolism and Inflammation in Cancer. Carcinogenesis 2020, 41, 405–416.
- Tanigawa, S.; Fujii, M.; Hou, D. Action of Nrf2 and Keap1 in ARE-Mediated NQO1 Expression by Quercetin. Free Radic. Biol. Med. 2007, 42, 1690–1703.
- Feng, R.-B.; Wang, Y.; He, C.; Yang, Y.; Wan, J.-B. Gallic Acid, a Natural Polyphenol, Protects against Tert-Butyl Hydroperoxide- Induced Hepatotoxicity by Activating ERK-Nrf2-Keap1-Mediated Antioxidative Response. Food Chem. Toxicol. 2018, 119, 479–488.
- Yang, M.; Jiang, Z.; Li, C.; Zhu, Y.; Li, Z.; Tang, Y.; Ni, C. Apigenin Prevents Metabolic Syndrome in High-Fructose Diet-Fed Mice by Keap1-Nrf2 Pathway. Biomed. Pharmacother. 2018, 105, 1283–1290.
- Lin, Y.-L.; Tsai, S.-H.; Lin-Shiau, S.-Y.; Ho, C.-T.; Lin, J.-K. Theaflavin-3,3′-Digallate from Black Tea Blocks the Nitric Oxide Synthase by down-Regulating the Activation of NF-ΚB in Macrophages. Eur. J. Pharmacol. 1999, 367, 379–388.
- Lin, Y.-L.; Lin, J.-K. (−)-Epigallocatechin-3-Gallate Blocks the Induction of Nitric Oxide Synthase by Down-Regulating Lipopolysaccharide-Induced Activity of Transcription Factor Nuclear Factor-ΚB. Mol. Pharmacol. 1997, 52, 465–472.
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox Homeostasis: The Golden Mean of Healthy Living. Redox Biol. 2016, 8, 205–215.
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How Do Nutritional Antioxidants Really Work: Nucleophilic Tone and Para-Hormesis versus Free Radical Scavenging in Vivo. Free Radic. Biol. Med. 2014, 66, 24–35.
- Watson, J. Oxidants, Antioxidants and the Current Incurability of Metastatic Cancers. Open Biol. 2013, 3, 120144.
- Wang, J.; Yi, J. Cancer Cell Killing via ROS: To Increase or Decrease, That Is the Question. Cancer Biol. Ther. 2008, 7, 1875–1884.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591.
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant Polyphenols in Cancer Treatment: Friend, Foe or Foil? Semin. Cancer Biol. 2017, 46, 1–13.
- Peixoto, P.; Cartron, P.-F.; Serandour, A.A.; Hervouet, E. From 1957 to Nowadays: A Brief History of Epigenetics. IJMS 2020, 21, 7571.
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating Genetic and Non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat. Rev. Genet. 2021, 22, 3–18.
- Darwiche, N. Epigenetic Mechanisms and the Hallmarks of Cancer: An Intimate Affair. Am. J. Cancer Res. 2020, 10, 1954–1978.
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027.
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521.
- Martire, S.; Banaszynski, L.A. The Roles of Histone Variants in Fine-Tuning Chromatin Organization and Function. Nat. Rev. Mol. Cell. Biol. 2020, 21, 522–541.
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.-Y.; Duarte, L.F.; Bernstein, E. Histone Variants: Emerging Players in Cancer Biology. Cell. Mol. Life Sci. 2014, 71, 379–404.
- Scaffidi, P. Histone H1 Alterations in Cancer. Biochim. Biophys. Acta 2016, 1859, 533–539.
- Lyubitelev, A.V.; Kirpichnikov, M.P.; Studitsky, V.M. The Role of Linker Histones in Carcinogenesis. Russ. J. Bioorg. Chem. 2021, 47, 278–287.
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. 2014, 9, 287–314.
- Zyner, K.G.; Simeone, A.; Flynn, S.M.; Doyle, C.; Marsico, G.; Adhikari, S.; Portella, G.; Tannahill, D.; Balasubramanian, S. G-Quadruplex DNA Structures in Human Stem Cells and Differentiation. Nat. Commun. 2022, 13, 142.
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-Gallate Reactivates Silenced Tumor Suppressor Genes, Cip1/P21 and P16INK4a, by Reducing DNA Methylation and Increasing Histones Acetylation in Human Skin Cancer Cells. Carcinogenesis 2011, 32, 537–544.
- Henning, S.M.; Wang, P.; Said, J.; Magyar, C.; Castor, B.; Doan, N.; Tosity, C.; Moro, A.; Gao, K.; Li, L.; et al. Polyphenols in Brewed Green Tea Inhibit Prostate Tumor Xenograft Growth by Localizing to the Tumor and Decreasing Oxidative Stress and Angiogenesis. J. Nutr. Biochem. 2012, 23, 1537–1542.
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-Epigallocatechin-3-Gallate Reverses the Expression of Various Tumor-Suppressor Genes by Inhibiting DNA Methyltransferases and Histone Deacetylases in Human Cervical Cancer Cells. Oncol. Rep. 2015, 33, 1976–1984.
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea Polyphenol (-)-Epigallocatechin-3-Gallate Inhibits DNA Methyltransferase and Reactivates Methylation-Silenced Genes in Cancer Cell Lines. Cancer Res. 2003, 63, 7563–7570.
- Jiang, A.; Wang, X.; Shan, X.; Li, Y.; Wang, P.; Jiang, P.; Feng, Q. Curcumin Reactivates Silenced Tumor Suppressor Gene RARβ by Reducing DNA Methylation. Phytother. Res. 2015, 29, 1237–1245.
- Du, L.; Xie, Z.; Wu, L.; Chiu, M.; Lin, J.; Chan, K.K.; Liu, S.; Liu, Z. Reactivation of RASSF1A in Breast Cancer Cells by Curcumin. Nutr. Cancer 2012, 64, 1228–1235.
- Kumar, U.; Sharma, U.; Rathi, G. Reversal of Hypermethylation and Reactivation of Glutathione S-Transferase Pi 1 Gene by Curcumin in Breast Cancer Cell Line. Tumour Biol. 2017, 39, 1010428317692258.
- Sharma, V.; Kumar, L.; Mohanty, S.K.; Maikhuri, J.P.; Rajender, S.; Gupta, G. Sensitization of Androgen Refractory Prostate Cancer Cells to Anti-Androgens through Re-Expression of Epigenetically Repressed Androgen Receptor-Synergistic Action of Quercetin and Curcumin. Mol. Cell. Endocrinol. 2016, 431, 12–23.
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-Based Combinatorial Resveratrol and Pterostilbene Alters DNA Damage Response by Affecting SIRT1 and DNMT Enzyme Expression, Including SIRT1-Dependent γ-H2AX and Telomerase Regulation in Triple-Negative Breast Cancer. BMC Cancer 2015, 15, 672.
- Medina-Aguilar, R.; Pérez-Plasencia, C.; Marchat, L.A.; Gariglio, P.; García Mena, J.; Rodríguez Cuevas, S.; Ruíz-García, E.; Astudillo-de la Vega, H.; Hernández Juárez, J.; Flores-Pérez, A.; et al. Methylation Landscape of Human Breast Cancer Cells in Response to Dietary Compound Resveratrol. PLoS ONE 2016, 11, e0157866.
- Zhu, W.; Qin, W.; Zhang, K.; Rottinghaus, G.E.; Chen, Y.-C.; Kliethermes, B.; Sauter, E.R. Trans-Resveratrol Alters Mammary Promoter Hypermethylation in Women at Increased Risk for Breast Cancer. Nutr. Cancer 2012, 64, 393–400.
- Majid, S.; Dar, A.A.; Ahmad, A.E.; Hirata, H.; Kawakami, K.; Shahryari, V.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Khatri, G.; et al. BTG3 Tumor Suppressor Gene Promoter Demethylation, Histone Modification and Cell Cycle Arrest by Genistein in Renal Cancer. Carcinogenesis 2009, 30, 662–670.
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein Reverses Hypermethylation and Induces Active Histone Modifications in Tumor Suppressor Gene B-Cell Translocation Gene 3 in Prostate Cancer. Cancer 2010, 116, 66–76.
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of Gene Methylation by Genistein or Lycopene in Breast Cancer Cells. Environ. Mol. Mutagen. 2008, 49, 36–45.
- Pandey, M.; Shukla, S.; Gupta, S. Promoter Demethylation and Chromatin Remodeling by Green Tea Polyphenols Leads to Re-Expression of GSTP1 in Human Prostate Cancer Cells. Int. J. Cancer 2010, 126, 2520–2533.
- Saldanha, S.N.; Kala, R.; Tollefsbol, T.O. Molecular Mechanisms for Inhibition of Colon Cancer Cells by Combined Epigenetic-Modulating Epigallocatechin Gallate and Sodium Butyrate. Exp. Cell. Res. 2014, 324, 40–53.
- Choi, K.-C.; Jung, M.G.; Lee, Y.-H.; Yoon, J.C.; Kwon, S.H.; Kang, H.-B.; Kim, M.-J.; Cha, J.-H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-Gallate, a Histone Acetyltransferase Inhibitor, Inhibits EBV-Induced B Lymphocyte Transformation via Suppression of RelA Acetylation. Cancer Res. 2009, 69, 583–592.
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; Rajasekaran, S.A. Curcumin-Induced HDAC Inhibition and Attenuation of Medulloblastoma Growth in Vitro and in Vivo. BMC Cancer 2011, 11, 144.
- Chen, Y.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, Both Histone Deacetylase and P300/CBP-Specific Inhibitor, Represses the Activity of Nuclear Factor Kappa B and Notch 1 in Raji Cells. Basic Clin. Pharm. Toxicol. 2007, 101, 427–433.
- Lee, W.-J.; Chen, Y.-R.; Tseng, T.-H. Quercetin Induces FasL-Related Apoptosis, in Part, through Promotion of Histone H3 Acetylation in Human Leukemia HL-60 Cells. Oncol. Rep. 2011, 25, 583–591.
- Xiao, X.; Shi, D.; Liu, L.; Wang, J.; Xie, X.; Kang, T.; Deng, W. Quercetin Suppresses Cyclooxygenase-2 Expression and Angiogenesis through Inactivation of P300 Signaling. PLoS ONE 2011, 6, e22934.
- Wang, R.-H.; Zheng, Y.; Kim, H.-S.; Xu, X.; Cao, L.; Luhasen, T.; Lee, M.-H.; Xiao, C.; Vassilopoulos, A.; Chen, W.; et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-Associated Tumorigenesis. Mol. Cell 2008, 32, 11–20.
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol Regulates PTEN/Akt Pathway through Inhibition of MTA1/HDAC Unit of the NuRD Complex in Prostate Cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 265–275.
- Kai, L.; Samuel, S.K.; Levenson, A.S. Resveratrol Enhances P53 Acetylation and Apoptosis in Prostate Cancer by Inhibiting MTA1/NuRD Complex. Int. J. Cancer 2010, 126, 1538–1548.
- Majid, S.; Kikuno, N.; Nelles, J.; Noonan, E.; Tanaka, Y.; Kawamoto, K.; Hirata, H.; Li, L.C.; Zhao, H.; Okino, S.T.; et al. Genistein Induces the P21WAF1/CIP1 and P16INK4a Tumor Suppressor Genes in Prostate Cancer Cells by Epigenetic Mechanisms Involving Active Chromatin Modification. Cancer Res. 2008, 68, 2736–2744.
- Li, Y.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic Regulation of Multiple Tumor-Related Genes Leads to Suppression of Breast Tumorigenesis by Dietary Genistein. PLoS ONE 2013, 8, e54369.
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51.
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17.
- Yan, H.; Bu, P. Non-Coding RNA in Cancer. Essays Biochem. 2021, 65, 625–639.
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-Coding RNAs in Oncology. Cell 2019, 179, 1033–1055.
- Tsang, W.P.; Kwok, T.T. Epigallocatechin Gallate Up-Regulation of MiR-16 and Induction of Apoptosis in Human Cancer Cells. J. Nutr. Biochem. 2010, 21, 140–146.
- Chakrabarti, M.; Khandkar, M.; Banik, N.L.; Ray, S.K. Alterations in Expression of Specific MicroRNAs by Combination of 4-HPR and EGCG Inhibited Growth of Human Malignant Neuroblastoma Cells. Brain Res. 2012, 1454, 1–13.
- Wang, H.; Bian, S.; Yang, C.S. Green Tea Polyphenol EGCG Suppresses Lung Cancer Cell Growth through Upregulating MiR-210 Expression Caused by Stabilizing HIF-1α. Carcinogenesis 2011, 32, 1881–1889.
- Zan, L.; Chen, Q.; Zhang, L.; Li, X. Epigallocatechin Gallate (EGCG) Suppresses Growth and Tumorigenicity in Breast Cancer Cells by Downregulation of MiR-25. Bioengineered 2019, 10, 374–382.
- Zhong, Z.; Dong, Z.; Yang, L.; Chen, X.; Gong, Z. Inhibition of Proliferation of Human Lung Cancer Cells by Green Tea Catechins Is Mediated by Upregulation of Let-7. Exp. Ther. Med. 2012, 4, 267–272.
- Appari, M.; Babu, K.R.; Kaczorowski, A.; Gross, W.; Herr, I. Sulforaphane, Quercetin and Catechins Complement Each Other in Elimination of Advanced Pancreatic Cancer by MiR-Let-7 Induction and K-Ras Inhibition. Int. J. Oncol. 2014, 45, 1391–1400.
- Nirgude, S.; Desai, S.; Choudhary, B. Curcumin Alters Distinct Molecular Pathways in Breast Cancer Subtypes Revealed by Integrated miRNA/mRNA Expression Analysis. Cancer Rep. 2022, 5, e1596.
- Wang, X.; Hang, Y.; Liu, J.; Hou, Y.; Wang, N.; Wang, M. Anticancer Effect of Curcumin Inhibits Cell Growth through MiR-21/PTEN/Akt Pathway in Breast Cancer Cell. Oncol. Lett. 2017, 13, 4825–4831.
- Liu, W.; Huang, M.; Zou, Q.; Lin, W. Curcumin Suppresses Gastric Cancer Biological Activity by Regulation of MiRNA-21: An in Vitro Study. Int. J. Clin. Exp. Pathol. 2018, 11, 5820–5829.
- Zhang, W.; Bai, W.; Zhang, W. MiR-21 Suppresses the Anticancer Activities of Curcumin by Targeting PTEN Gene in Human Non-Small Cell Lung Cancer A549 Cells. Clin. Transl. Oncol. 2014, 16, 708–713.
- Mudduluru, G.; George-William, J.N.; Muppala, S.; Asangani, I.A.; Kumarswamy, R.; Nelson, L.D.; Allgayer, H. Curcumin Regulates MiR-21 Expression and Inhibits Invasion and Metastasis in Colorectal Cancer. Biosci. Rep. 2011, 31, 185–197.
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P.N. Difluorinated-Curcumin (CDF) Restores PTEN Expression in Colon Cancer Cells by down-Regulating MiR-21. PLoS ONE 2013, 8, e68543.
- Yang, C.H.; Yue, J.; Sims, M.; Pfeffer, L.M. The Curcumin Analog EF24 Targets NF-ΚB and MiRNA-21, and Has Potent Anticancer Activity in Vitro and in Vivo. PLoS ONE 2013, 8, e71130.
- Li, X.; Xie, W.; Xie, C.; Huang, C.; Zhu, J.; Liang, Z.; Deng, F.; Zhu, M.; Zhu, W.; Wu, R.; et al. Curcumin Modulates MiR-19/PTEN/AKT/P53 Axis to Suppress Bisphenol A-Induced MCF-7 Breast Cancer Cell Proliferation: CURCUMIN MODULATES BPA-DYSREGULATED MIR-19/PTEN/AKT/P53 AXIS. Phytother. Res. 2014, 28, 1553–1560.
- Sun, M.; Estrov, Z.; Ji, Y.; Coombes, K.R.; Harris, D.H.; Kurzrock, R. Curcumin (Diferuloylmethane) Alters the Expression Profiles of MicroRNAs in Human Pancreatic Cancer Cells. Mol. Cancer Ther. 2008, 7, 464–473.
- Zhang, J.; Zhang, T.; Ti, X.; Shi, J.; Wu, C.; Ren, X.; Yin, H. Curcumin Promotes Apoptosis in A549/DDP Multidrug-Resistant Human Lung Adenocarcinoma Cells through an MiRNA Signaling Pathway. Biochem. Biophys. Res. Commun. 2010, 399, 1–6.
- Zhu, M.; Zheng, Z.; Huang, J.; Ma, X.; Huang, C.; Wu, R.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; et al. Modulation of MiR-34a in Curcumin-Induced Antiproliferation of Prostate Cancer Cells. J. Cell. Biochem. 2019, 120, 15616–15624.
- Liu, W.-L.; Chang, J.-M.; Chong, I.-W.; Hung, Y.-L.; Chen, Y.-H.; Huang, W.-T.; Kuo, H.-F.; Hsieh, C.-C.; Liu, P.-L. Curcumin Inhibits LIN-28A through the Activation of MiRNA-98 in the Lung Cancer Cell Line A549. Molecules 2017, 22, 929.
- Zhao, J.; Fang, Z.; Zha, Z.; Sun, Q.; Wang, H.; Sun, M.; Qiao, B. Quercetin Inhibits Cell Viability, Migration and Invasion by Regulating MiR-16/HOXA10 Axis in Oral Cancer. Eur. J. Pharmacol. 2019, 847, 11–18.
- Sonoki, H.; Sato, T.; Endo, S.; Matsunaga, T.; Yamaguchi, M.; Yamazaki, Y.; Sugatani, J.; Ikari, A. Quercetin Decreases Claudin-2 Expression Mediated by Up-Regulation of MicroRNA MiR-16 in Lung Adenocarcinoma A549 Cells. Nutrients 2015, 7, 4578–4592.
- Lou, G.; Liu, Y.; Wu, S.; Xue, J.; Yang, F.; Fu, H.; Zheng, M.; Chen, Z. The P53/MiR-34a/SIRT1 Positive Feedback Loop in Quercetin-Induced Apoptosis. Cell. Physiol. Biochem. 2015, 35, 2192–2202.
- Tao, S.; He, H.; Chen, Q. Quercetin Inhibits Proliferation and Invasion Acts by Up-Regulating MiR-146a in Human Breast Cancer Cells. Mol. Cell. Biochem. 2015, 402, 93–100.
- Pratheeshkumar, P.; Son, Y.-O.; Divya, S.P.; Wang, L.; Turcios, L.; Roy, R.V.; Hitron, J.A.; Kim, D.; Dai, J.; Asha, P.; et al. Quercetin Inhibits Cr(VI)-Induced Malignant Cell Transformation by Targeting MiR-21-PDCD4 Signaling Pathway. Oncotarget 2017, 8, 52118–52131.
- Nwaeburu, C.C.; Bauer, N.; Zhao, Z.; Abukiwan, A.; Gladkich, J.; Benner, A.; Herr, I. Up-Regulation of MicroRNA Let-7c by Quercetin Inhibits Pancreatic Cancer Progression by Activation of Numbl. Oncotarget 2016, 7, 58367–58380.
- Del Follo-Martinez, A.; Banerjee, N.; Li, X.; Safe, S.; Mertens-Talcott, S. Resveratrol and Quercetin in Combination Have Anticancer Activity in Colon Cancer Cells and Repress Oncogenic MicroRNA-27a. Nutr. Cancer 2013, 65, 494–504.
- Wang, G.; Dai, F.; Yu, K.; Jia, Z.; Zhang, A.; Huang, Q.; Kang, C.; Jiang, H.; Pu, P. Resveratrol Inhibits Glioma Cell Growth via Targeting Oncogenic MicroRNAs and Multiple Signaling Pathways. Int. J. Oncol. 2015, 46, 1739–1747.
- Li, H.; Jia, Z.; Li, A.; Jenkins, G.; Yang, X.; Hu, J.; Guo, W. Resveratrol Repressed Viability of U251 Cells by MiR-21 Inhibiting of NF-ΚB Pathway. Mol. Cell. Biochem. 2013, 382, 137–143.
- Zhou, C.; Ding, J.; Wu, Y. Resveratrol Induces Apoptosis of Bladder Cancer Cells via MiR-21 Regulation of the Akt/Bcl-2 Signaling Pathway. Mol. Med. Rep. 2014, 9, 1467–1473.
- Sheth, S.; Jajoo, S.; Kaur, T.; Mukherjea, D.; Sheehan, K.; Rybak, L.P.; Ramkumar, V. Resveratrol Reduces Prostate Cancer Growth and Metastasis by Inhibiting the Akt/MicroRNA-21 Pathway. PLoS ONE 2012, 7, e51655.
- Wang, H.; Feng, H.; Zhang, Y. Resveratrol Inhibits Hypoxia-Induced Glioma Cell Migration and Invasion by the p-STAT3/MiR-34a Axis. Neoplasma 2016, 63, 532–539.
- Yang, S.; Li, W.; Sun, H.; Wu, B.; Ji, F.; Sun, T.; Chang, H.; Shen, P.; Wang, Y.; Zhou, D. Resveratrol Elicits Anti-Colorectal Cancer Effect by Activating MiR-34c-KITLG in Vitro and in Vivo. BMC Cancer 2015, 15, 969.
- Venkatadri, R.; Muni, T.; Iyer, A.K.V.; Yakisich, J.S.; Azad, N. Role of Apoptosis-Related MiRNAs in Resveratrol-Induced Breast Cancer Cell Death. Cell Death Dis. 2016, 7, e2104.
- Sun, Q.; Cong, R.; Yan, H.; Gu, H.; Zeng, Y.; Liu, N.; Chen, J.; Wang, B. Genistein Inhibits Growth of Human Uveal Melanoma Cells and Affects MicroRNA-27a and Target Gene Expression. Oncol. Rep. 2009, 22, 563–567.
- Xu, L.; Xiang, J.; Shen, J.; Zou, X.; Zhai, S.; Yin, Y.; Li, P.; Wang, X.; Sun, Q. Oncogenic MicroRNA-27a Is a Target for Genistein in Ovarian Cancer Cells. Anticancer Agents Med. Chem. 2013, 13, 1126–1132.
- Yang, Y.; Zang, A.; Jia, Y.; Shang, Y.; Zhang, Z.; Ge, K.; Zhang, J.; Fan, W.; Wang, B. Genistein Inhibits A549 Human Lung Cancer Cell Proliferation via MiR-27a and MET Signaling. Oncol. Lett. 2016, 12, 2189–2193.
- Avci, C.B.; Susluer, S.Y.; Caglar, H.O.; Balci, T.; Aygunes, D.; Dodurga, Y.; Gunduz, C. Genistein-Induced Mir-23b Expression Inhibits the Growth of Breast Cancer Cells. Contemp. Oncol. 2015, 19, 32–35.
- Chiyomaru, T.; Yamamura, S.; Zaman, M.S.; Majid, S.; Deng, G.; Shahryari, V.; Saini, S.; Hirata, H.; Ueno, K.; Chang, I.; et al. Genistein Suppresses Prostate Cancer Growth through Inhibition of Oncogenic MicroRNA-151. PLoS ONE 2012, 7, e43812.
- De la Parra, C.; Castillo-Pichardo, L.; Cruz-Collazo, A.; Cubano, L.; Redis, R.; Calin, G.A.; Dharmawardhane, S. Soy Isoflavone Genistein-Mediated Downregulation of MiR-155 Contributes to the Anticancer Effects of Genistein. Nutr. Cancer 2016, 68, 154–164.
- Xia, J.; Duan, Q.; Ahmad, A.; Bao, B.; Banerjee, S.; Shi, Y.; Ma, J.; Geng, J.; Chen, Z.; Rahman, K.M.W.; et al. Genistein Inhibits Cell Growth and Induces Apoptosis through Up-Regulation of MiR-34a in Pancreatic Cancer Cells. Curr. Drug Targets 2012, 13, 1750–1756.
- Chiyomaru, T.; Yamamura, S.; Fukuhara, S.; Yoshino, H.; Kinoshita, T.; Majid, S.; Saini, S.; Chang, I.; Tanaka, Y.; Enokida, H.; et al. Genistein Inhibits Prostate Cancer Cell Growth by Targeting MiR-34a and Oncogenic HOTAIR. PLoS ONE 2013, 8, e70372.
- Mishra, S.; Verma, S.S.; Rai, V.; Awasthee, N.; Chava, S.; Hui, K.M.; Kumar, A.P.; Challagundla, K.B.; Sethi, G.; Gupta, S.C. Long Non-Coding RNAs Are Emerging Targets of Phytochemicals for Cancer and Other Chronic Diseases. Cell. Mol. Life Sci. 2019, 76, 1947–1966.
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367.
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435.
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41.
- Mitchell, J.P.; Carmody, R.J. NF-ΚB and the Transcriptional Control of Inflammation. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 335, pp. 41–84. ISBN 978-0-12-812339-3.
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK Biology. Immunol. Rev. 2012, 246, 239–253.
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85.
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. WIREs Mech. Dis. 2016, 8, 227–241.
- Disis, M.L. Immune Regulation of Cancer. J. Clin. Oncol. 2010, 28, 4531–4538.
- Laveti, D.; Kumar, M.; Hemalatha, R.; Sistla, R.; Naidu, V.; Talla, V.; Verma, V.; Kaur, N.; Nagpal, R. Anti-Inflammatory Treatments for Chronic Diseases: A Review. Inflamm. Allergy Drug Targets 2013, 12, 349–361.
- Xie, T.-X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-KappaB Activity Regulates the Expression of VEGF and IL-8 and Tumor Angiogenesis of Human Glioblastoma. Oncol. Rep. 2010, 23, 725–732.
- Kumar, S.; Mehta, K. Tissue Transglutaminase Constitutively Activates HIF-1α Promoter and Nuclear Factor-ΚB via a Non-Canonical Pathway. PLoS ONE 2012, 7, e49321.
- Peng, G.; Dixon, D.A.; Muga, S.J.; Smith, T.J.; Wargovich, M.J. Green Tea Polyphenol (−)-Epigallocatechin-3-Gallate Inhibits Cyclooxygenase-2 Expression in Colon Carcinogenesis. Mol. Carcinog. 2006, 45, 309–319.
- Chen, P.C.; Wheeler, D.S.; Malhotra, V.; Odoms, K.; Denenberg, A.G.; Wong, H.R. A Green Tea-Derived Polyphenol, Epigallocatechin-3-Gallate, Inhibits IkappaB Kinase Activation and IL-8 Gene Expression in Respiratory Epithelium. Inflammation 2002, 26, 233–241.
- Wheeler, D.S.; Catravas, J.D.; Odoms, K.; Denenberg, A.; Malhotra, V.; Wong, H.R. Epigallocatechin-3-Gallate, a Green Tea–Derived Polyphenol, Inhibits IL-1β-Dependent Proinflammatory Signal Transduction in Cultured Respiratory Epithelial Cells. J. Nutr. 2004, 134, 1039–1044.
- Yang, F.; Oz, H.S.; Barve, S.; de Villiers, W.J.; McClain, C.J.; Varilek, G.W. The Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate Blocks Nuclear Factor-Kappa B Activation by Inhibiting I Kappa B Kinase Activity in the Intestinal Epithelial Cell Line IEC-6. Mol. Pharmacol. 2001, 60, 528–533.
- Fechtner, S.; Singh, A.; Chourasia, M.; Ahmed, S. Molecular Insights into the Differences in Anti-Inflammatory Activities of Green Tea Catechins on IL-1β Signaling in Rheumatoid Arthritis Synovial Fibroblasts. Toxicol. Appl. Pharmacol. 2017, 329, 112–120.
- Singh, A.K.; Umar, S.; Riegsecker, S.; Chourasia, M.; Ahmed, S. Regulation of Transforming Growth Factor β-Activated Kinase Activation by Epigallocatechin-3-Gallate in Rheumatoid Arthritis Synovial Fibroblasts: Suppression of K63 -Linked Autoubiquitination of Tumor Necrosis Factor Receptor-Associated Factor 6: TAK1 INHIBITION BY EGCG IN RA. Arthritis Rheumatol. 2016, 68, 347–358.
- Varthya, S.B.; Sarma, P.; Bhatia, A.; Shekhar, N.; Prajapat, M.; Kaur, H.; Thangaraju, P.; Kumar, S.; Singh, R.; Siingh, A.; et al. Efficacy of Green Tea, Its Polyphenols and Nanoformulation in Experimental Colitis and the Role of Non-Canonical and Canonical Nuclear Factor Kappa Beta (NF-KB) Pathway: A Preclinical in-Vivo and in-Silico Exploratory Study. J. Biomol. Struct. Dyn. 2021, 39, 5314–5326.
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. The Tea Catechin Epigallocatechin Gallate Inhibits NF-ΚB-Mediated Transcriptional Activation by Covalent Modification. Arch. Biochem. Biophys. 2020, 695, 108620.
- Bachmeier, B.E.; Mohrenz, I.V.; Mirisola, V.; Schleicher, E.; Romeo, F.; Höhneke, C.; Jochum, M.; Nerlich, A.G.; Pfeffer, U. Curcumin Downregulates the Inflammatory Cytokines CXCL1 and -2 in Breast Cancer Cells via NFκB. Carcinogenesis 2008, 29, 779–789.
- Olivera, A.; Moore, T.W.; Hu, F.; Brown, A.P.; Sun, A.; Liotta, D.C.; Snyder, J.P.; Yoon, Y.; Shim, H.; Marcus, A.I.; et al. Inhibition of the NF-ΚB Signaling Pathway by the Curcumin Analog, 3,5-Bis(2-Pyridinylmethylidene)-4-Piperidone (EF31): Anti-Inflammatory and Anti-Cancer Properties. Int. Immunopharmacol. 2012, 12, 368–377.
- Abdel-Hakeem, M.A.; Mongy, S.; Hassan, B.; Tantawi, O.I.; Badawy, I. Curcumin Loaded Chitosan-Protamine Nanoparticles Revealed Antitumor Activity via Suppression of NF-ΚB, Proinflammatory Cytokines and Bcl-2 Gene Expression in the Breast Cancer Cells. J. Pharm. Sci. 2021, 110, 3298–3305.
- Coker-Gurkan, A.; Bulut, D.; Genc, R.; Arisan, E.-D.; Obakan-Yerlikaya, P.; Palavan-Unsal, N. Curcumin Prevented Human Autocrine Growth Hormone (GH) Signaling Mediated NF-ΚB Activation and MiR-183-96-182 Cluster Stimulated Epithelial Mesenchymal Transition in T47D Breast Cancer Cells. Mol. Biol. Rep. 2019, 46, 355–369.
- Ghasemi, F.; Shafiee, M.; Banikazemi, Z.; Pourhanifeh, M.H.; Khanbabaei, H.; Shamshirian, A.; Amiri Moghadam, S.; ArefNezhad, R.; Sahebkar, A.; Avan, A.; et al. Curcumin Inhibits NF-KB and Wnt/β-Catenin Pathways in Cervical Cancer Cells. Pathol.-Res. Pract. 2019, 215, 152556.
- Tian, F.; Zhang, C.; Tian, W.; Jiang, Y.; Zhang, X. Comparison of the Effect of P65 SiRNA and Curcumin in Promoting Apoptosis in Esophageal Squamous Cell Carcinoma Cells and in Nude Mice. Oncol. Rep. 2012, 28, 232–240.
- Mu, Y.-T.; Feng, H.-H.; Yu, J.-Q.; Liu, Z.-K.; Wang, Y.; Shao, J.; Li, R.-H.; Li, D.-K. Curcumin Suppressed Proliferation and Migration of Human Retinoblastoma Cells through Modulating NF-ΚB Pathway. Int. Ophthalmol. 2020, 40, 2435–2440.
- Han, M.; Song, Y.; Zhang, X. Quercetin Suppresses the Migration and Invasion in Human Colon Cancer Caco-2 Cells Through Regulating Toll-like Receptor 4/Nuclear Factor-Kappa B Pathway. Pharmacogn. Mag. 2016, 12, S237–S244.
- Zhang, X.-A.; Zhang, S.; Yin, Q.; Zhang, J. Quercetin Induces Human Colon Cancer Cells Apoptosis by Inhibiting the Nuclear Factor-Kappa B Pathway. Pharmacogn. Mag. 2015, 11, 404–409.
- Youn, H.; Jeong, J.-C.; Jeong, Y.S.; Kim, E.-J.; Um, S.-J. Quercetin Potentiates Apoptosis by Inhibiting Nuclear Factor-KappaB Signaling in H460 Lung Cancer Cells. Biol. Pharm. Bull. 2013, 36, 944–951.
- Lai, W.-W.; Hsu, S.-C.; Chueh, F.-S.; Chen, Y.-Y.; Yang, J.-S.; Lin, J.-P.; Lien, J.-C.; Tsai, C.-H.; Chung, J.-G. Quercetin Inhibits Migration and Invasion of SAS Human Oral Cancer Cells through Inhibition of NF-ΚB and Matrix Metalloproteinase-2/-9 Signaling Pathways. Anticancer. Res. 2013, 33, 1941–1950.
- Zhang, W.; Yin, G.; Dai, J.; Sun, Y.; Hoffman, R.M.; Yang, Z.; Fan, Y. Chemoprevention by Quercetin of Oral Squamous Cell Carcinoma by Suppression of the NF-ĸB Signaling Pathway in DMBA-Treated Hamsters. Anticancer. Res. 2017, 37, 4041–4049.
- Shree, A.; Islam, J.; Sultana, S. Quercetin Ameliorates Reactive Oxygen Species Generation, Inflammation, Mucus Depletion, Goblet Disintegration, and Tumor Multiplicity in Colon Cancer: Probable Role of Adenomatous Polyposis Coli, β-catenin. Phytother. Res. 2021, 35, 2171–2184.
- García-Zepeda, S.P.; García-Villa, E.; Díaz-Chávez, J.; Hernández-Pando, R.; Gariglio, P. Resveratrol Induces Cell Death in Cervical Cancer Cells through Apoptosis and Autophagy. Eur. J. Cancer Prev. 2013, 22, 577–584.
- Dariya, B.; Behera, S.K.; Srivani, G.; Farran, B.; Alam, A.; Nagaraju, G.P. Computational Analysis of Nuclear Factor-ΚB and Resveratrol in Colorectal Cancer. J. Biomol. Struct. Dyn. 2021, 39, 2914–2922.
- Wu, F.; Cui, L. Resveratrol Suppresses Melanoma by Inhibiting NF-ΚB/MiR-221 and Inducing TFG Expression. Arch. Derm. Res. 2017, 309, 823–831.
- Qian, W.; Xiao, Q.; Wang, L.; Qin, T.; Xiao, Y.; Li, J.; Yue, Y.; Zhou, C.; Duan, W.; Ma, Q.; et al. Resveratrol Slows the Tumourigenesis of Pancreatic Cancer by Inhibiting NFκB Activation. Biomed. Pharmacother. 2020, 127, 110116.
- Rawat, D.; Chhonker, S.K.; Naik, R.A.; Koiri, R.K. Modulation of Antioxidant Enzymes, SIRT1 and NF-κB by Resveratrol and Nicotinamide in Alcohol-aflatoxin B1-induced Hepatocellular Carcinoma. J. Biochem. Mol. Toxicol. 2021, 35, e22625.
- Tino, A.B.; Chitcholtan, K.; Sykes, P.H.; Garrill, A. Resveratrol and Acetyl-Resveratrol Modulate Activity of VEGF and IL-8 in Ovarian Cancer Cell Aggregates via Attenuation of the NF-ΚB Protein. J. Ovarian Res. 2016, 9, 84.
- Fukui, M.; Yamabe, N.; Kang, K.S.; Zhu, B.T. Growth-Stimulatory Effect of Resveratrol in Human Cancer Cells. Mol. Carcinog. 2010, 49, 750–759.
- Xie, J.; Wang, J.; Zhu, B. Genistein Inhibits the Proliferation of Human Multiple Myeloma Cells through Suppression of Nuclear Factor-ΚB and Upregulation of MicroRNA-29b. Mol. Med. Rep. 2016, 13, 1627–1632.
- Ozturk, S.; Alp, E.; Saglam, A.Y.; Konac, E.; Menevse, E. The Effects of Thymoquinone and Genistein Treatment on Telomerase Activity, Apoptosis, Angiogenesis, and Survival in Thyroid Cancer Cell Lines. J. Can. Res. Ther. 2017, 14, 328–334.
- Zhang, J.; Su, H.; Li, Q.; Li, J.; Zhao, Q. Genistein Decreases A549 Cell Viability via Inhibition of the PI3K/AKT/HIF-1α/VEGF and NF-ΚB/COX-2 Signaling Pathways. Mol. Med. Rep. 2017, 15, 2296–2302.
- Zhou, P.; Wang, C.; Hu, Z.; Chen, W.; Qi, W.; Li, A. Genistein Induces Apoptosis of Colon Cancer Cells by Reversal of Epithelial-to-Mesenchymal via a Notch1/NF-ΚB/Slug/E-Cadherin Pathway. BMC Cancer 2017, 17, 813.
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein Inhibits MDA-MB-231 Triple-Negative Breast Cancer Cell Growth by Inhibiting NF-ΚB Activity via the Notch-1 Pathway. Int. J. Mol. Med. 2012, 30, 337–343.
- Luo, Y.; Wang, S.; Zhou, Z.; Wang, Z.; Zhang, Y.; Zhang, Y.; Zhao, P. Apoptotic Effect of Genistein on Human Colon Cancer Cells via Inhibiting the Nuclear Factor-Kappa B (NF-ΚB) Pathway. Tumor Biol. 2014, 35, 11483–11488.
- Vilela, F.M.P.; Syed, D.N.; Chamcheu, J.C.; Calvo-Castro, L.A.; Fortes, V.S.; Fonseca, M.J.V.; Mukhtar, H. Biotransformed Soybean Extract (BSE) Inhibits Melanoma Cell Growth and Viability in Vitro: Involvement of Nuclear Factor-Kappa B Signaling. PLoS ONE 2014, 9, e103248.
- Alorda-Clara, M.; Torrens-Mas, M.; Morla-Barcelo, P.M.; Roca, P.; Sastre-Serra, J.; Pons, D.G.; Oliver, J. High Concentrations of Genistein Decrease Cell Viability Depending on Oxidative Stress and Inflammation in Colon Cancer Cell Lines. IJMS 2022, 23, 7526.
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901.
- Ambati, G.G.; Jachak, S.M. Natural Product Inhibitors of Cyclooxygenase (COX) Enzyme: A Review on Current Status and Future Perspectives. Curr. Med. Chem. 2021, 28, 1877–1905.
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618.
- Barrett, K.E.; Wu, G.D. Influence of the Microbiota on Host Physiology-Moving beyond the Gut: Editorial. J. Physiol. 2017, 595, 433–435.
- Young, V.B. The Role of the Microbiome in Human Health and Disease: An Introduction for Clinicians. BMJ 2017, 356, j831.
- LaCourse, K.D.; Johnston, C.D.; Bullman, S. The Relationship between Gastrointestinal Cancers and the Microbiota. Lancet Gastroenterol. Hepatol. 2021, 6, 498–509.
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA. Science 2019, 363, eaar7785.
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni Promotes Colorectal Tumorigenesis through the Action of Cytolethal Distending Toxin. Gut 2019, 68, 289–300.
- Hashemi Goradel, N.; Heidarzadeh, S.; Jahangiri, S.; Farhood, B.; Mortezaee, K.; Khanlarkhani, N.; Negahdari, B. Fusobacterium nucleatum and Colorectal Cancer: A Mechanistic Overview. J. Cell. Physiol. 2019, 234, 2337–2344.
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori Virulence Factors—Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells 2020, 10, 27.
- Flukes, S.L.; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer. Schistosomes, Liver Flukes and Helicobacter pylori: This Publication Represents the Views and Expert Opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Which Met in Lyon, 7–14 June 1994; International Agency for Research on Cancer, Ed.; IARC monographs on the evaluation of carcinogenic risks to humans; IARC: Lyon, France, 1994; ISBN 978-92-832-1261-4.
- Fang, Y.; Yan, C.; Zhao, Q.; Xu, J.; Liu, Z.; Gao, J.; Zhu, H.; Dai, Z.; Wang, D.; Tang, D. The Roles of Microbial Products in the Development of Colorectal Cancer: A Review. Bioengineered 2021, 12, 720–735.
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689.
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-Omics to Mechanism? Trends Microbiol. 2020, 28, 401–423.
- Daglia, M. Polyphenols as Antimicrobial Agents. Curr. Opin. Biotechnol. 2012, 23, 174–181.
- Bittencourt, M.L.F.; Rodrigues, R.P.; Kitagawa, R.R.; Gonçalves, R.D.C.R. The Gastroprotective Potential of Silibinin against Helicobacter pylori Infection and Gastric Tumor Cells. Life Sci. 2020, 256, 117977.
- Escandón, R.A.; del Campo, M.; López-Solis, R.; Obreque-Slier, E.; Toledo, H. Antibacterial Effect of Kaempferol and (−)-Epicatechin on Helicobacter pylori. Eur. Food Res. Technol. 2016, 242, 1495–1502.
- Martini, S.; D’Addario, C.; Colacevich, A.; Focardi, S.; Borghini, F.; Santucci, A.; Figura, N.; Rossi, C. Antimicrobial Activity against Helicobacter pylori Strains and Antioxidant Properties of Blackberry Leaves (Rubus ulmifolius) and Isolated Compounds. Int. J. Antimicrob. Agents 2009, 34, 50–59.
- Chen, M.; Su, C.; Yang, J.; Lu, C.; Hou, Y.; Wu, J.; Hsu, Y. Baicalin, Baicalein, and Lactobacillus Rhamnosus JB3 Alleviated Helicobacter pylori Infections in Vitro and in Vivo. J. Food Sci. 2018, 83, 3118–3125.
- Mandalari, G.; Bisignano, C.; Cirmi, S.; Navarra, M. Effectiveness of Citrus Fruits on Helicobacter pylori. Evid.-Based Complement Altern. Med. 2017, 2017, 8379262.
- Park, J.U.; Cho, J.S.; Kim, J.S.; Kim, H.K.; Jo, Y.H.; Rahman, M.A.A.; Lee, Y.I. Synergistic Effect of Rubus crataegifolius and Ulmus macrocarpa against Helicobacter pylori Clinical Isolates and Gastritis. Front. Pharmacol. 2020, 11, 4.
- Ranilla, L.G.; Apostolidis, E.; Shetty, K. Antimicrobial Activity of an Amazon Medicinal Plant (Chancapiedra) (Phyllanthus niruri L.) against Helicobacter pylori and Lactic Acid Bacteria: Phyllanthus niruri L. and antimicrobial activity. Phytother. Res. 2012, 26, 791–799.
- Kareem, S.M.; Mahmood, S.S.; Hindi, N.K. Effects of Curcumin and Silymarin on the Shigella dysenteriae and Campylobacter jejuni In Vitro. J. Gastrointest. Cancer 2020, 51, 824–828.
- Castillo, S.; Heredia, N.; García, S. 2(5H)-Furanone, Epigallocatechin Gallate, and a Citric-Based Disinfectant Disturb Quorum-Sensing Activity and Reduce Motility and Biofilm Formation of Campylobacter jejuni. Folia Microbiol. 2015, 60, 89–95.
- Lobo de Sá, F.D.; Heimesaat, M.M.; Bereswill, S.; Nattramilarasu, P.K.; Schulzke, J.D.; Bücker, R. Resveratrol Prevents Campylobacter jejuni-Induced Leaky Gut by Restoring Occludin and Claudin-5 in the Paracellular Leak Pathway. Front. Pharmacol. 2021, 12, 640572.
- Heimesaat, M.M.; Mousavi, S.; Escher, U.; Lobo de Sá, F.D.; Peh, E.; Schulzke, J.-D.; Kittler, S.; Bücker, R.; Bereswill, S. Resveratrol Alleviates Acute Campylobacter jejuni Induced Enterocolitis in a Preclinical Murine Intervention Study. Microorganisms 2020, 8, 1858.
- Ben Lagha, A.; Haas, B.; Grenier, D. Tea Polyphenols Inhibit the Growth and Virulence Properties of Fusobacterium nucleatum. Sci. Rep. 2017, 7, 44815.
- Lim, Y.R.I.; Preshaw, P.M.; Lim, L.P.; Ong, M.M.A.; Lin, H.-S.; Tan, K.S. Pterostilbene Complexed with Cyclodextrin Exerts Antimicrobial and Anti-Inflammatory Effects. Sci. Rep. 2020, 10, 9072.
- Andrade, F.D.O.; Liu, F.; Zhang, X.; Rosim, M.P.; Dani, C.; Cruz, I.; Wang, T.T.Y.; Helferich, W.; Li, R.W.; Hilakivi-Clarke, L. Genistein Reduces the Risk of Local Mammary Cancer Recurrence and Ameliorates Alterations in the Gut Microbiota in the Offspring of Obese Dams. Nutrients 2021, 13, 201.
- Fernández, J.; García, L.; Monte, J.; Villar, C.; Lombó, F. Functional Anthocyanin-Rich Sausages Diminish Colorectal Cancer in an Animal Model and Reduce Pro-Inflammatory Bacteria in the Intestinal Microbiota. Genes 2018, 9, 133.
- McFadden, R.-M.T.; Larmonier, C.B.; Shehab, K.W.; Midura-Kiela, M.; Ramalingam, R.; Harrison, C.A.; Besselsen, D.G.; Chase, J.H.; Caporaso, J.G.; Jobin, C.; et al. The Role of Curcumin in Modulating Colonic Microbiota during Colitis and Colon Cancer Prevention. Inflamm. Bowel Dis. 2015, 21, 2483–2494.
- Chen, L.; Jiang, B.; Zhong, C.; Guo, J.; Zhang, L.; Mu, T.; Zhang, Q.; Bi, X. Chemoprevention of Colorectal Cancer by Black Raspberry Anthocyanins Involved the Modulation of Gut Microbiota and SFRP2 Demethylation. Carcinogenesis 2018, 39, 471–481.
- Wu, J.-C.; Tsai, M.-L.; Lai, C.-S.; Lo, C.-Y.; Ho, C.-T.; Wang, Y.-J.; Pan, M.-H. Polymethoxyflavones Prevent BenzoPyrene/Dextran Sodium Sulfate-Induced Colorectal Carcinogenesis through Modulating Xenobiotic Metabolism and Ameliorate Autophagic Defect in ICR Mice: PMFs Prevent B a P/DSS-Induced Colorectal Carcinogenesis. Int. J. Cancer 2018, 142, 1689–1701.
- Wu, M.; Wu, Y.; Deng, B.; Li, J.; Cao, H.; Qu, Y.; Qian, X.; Zhong, G. Isoliquiritigenin Decreases the Incidence of Colitis-Associated Colorectal Cancer by Modulating the Intestinal Microbiota. Oncotarget 2016, 7, 85318–85331.
- Gong, Y.; Dong, R.; Gao, X.; Li, J.; Jiang, L.; Zheng, J.; Cui, S.; Ying, M.; Yang, B.; Cao, J.; et al. Neohesperidin Prevents Colorectal Tumorigenesis by Altering the Gut Microbiota. Pharmacol. Res. 2019, 148, 104460.
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-Associated and Non-Tumour-Associated Microbiota in Colorectal Cancer. Gut 2017, 66, 633–643.
- Wang, X.; Ye, T.; Chen, W.-J.; Lv, Y.; Hao, Z.; Chen, J.; Zhao, J.-Y.; Wang, H.-P.; Cai, Y.-K. Structural shift of gut microbiota during chemo-preventive effects of epigallocatechin gallate on colorectal carcinogenesis in mice. World J. Gastroenterol. 2017, 23, 8128–8139.
- Messaoudene, M.; Pidgeon, R.; Richard, C.; Ponce, M.; Diop, K.; Benlaifaoui, M.; Nolin-Lapalme, A.; Cauchois, F.; Malo, J.; Belkaid, W.; et al. A Natural Polyphenol Exerts Antitumor Activity and Circumvents Anti–PD-1 Resistance through Effects on the Gut Microbiota. Cancer Discov. 2022, 12, 1070–1087.
- Jain, N. The Early Life Education of the Immune System: Moms, Microbes and (Missed) Opportunities. Gut Microbes 2020, 12, 1824564.
- Minarrieta, L.; Ghorbani, P.; Sparwasser, T.; Berod, L. Metabolites: Deciphering the Molecular Language between DCs and Their Environment. Semin. Immunopathol. 2017, 39, 177–198.
- Suzuki, H.; Jeong, K.I.; Itoh, K.; Doi, K. Regional Variations in the Distributions of Small Intestinal Intraepithelial Lymphocytes in Germ-Free and Specific Pathogen-Free Mice. Exp. Mol. Pathol. 2002, 72, 230–235.
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. Individual Intestinal Symbionts Induce a Distinct Population of RORγ+ Regulatory T Cells. Science 2015, 349, 993–997.
- Ramanan, D.; Sefik, E.; Galván-Peña, S.; Wu, M.; Yang, L.; Yang, Z.; Kostic, A.; Golovkina, T.V.; Kasper, D.L.; Mathis, D.; et al. An Immunologic Mode of Multigenerational Transmission Governs a Gut Treg Setpoint. Cell 2020, 181, 1276–1290.e13.
- Mandaliya, D.K.; Patel, S.; Seshadri, S. The Combinatorial Effect of Acetate and Propionate on High-Fat Diet Induced Diabetic Inflammation or Metaflammation and T Cell Polarization. Inflammation 2021, 44, 68–79.
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573.
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature 2019, 576, 143–148.
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial Bile Acid Metabolites Modulate Gut RORγ+ Regulatory T Cell Homeostasis. Nature 2020, 577, 410–415.
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The Role of Tumor Microenvironment in Therapeutic Resistance. Oncotarget 2017, 8, 3933–3945.
- Prager, I.; Watzl, C. Mechanisms of Natural Killer Cell-Mediated Cellular Cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329.
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056.
- Kroemer, G.; Zitvogel, L. The Breakthrough of the Microbiota. Nat. Rev. Immunol. 2018, 18, 87–88.
- Focaccetti, C.; Izzi, V.; Benvenuto, M.; Fazi, S.; Ciuffa, S.; Giganti, M.G.; Potenza, V.; Manzari, V.; Modesti, A.; Bei, R. Polyphenols as Immunomodulatory Compounds in the Tumor Microenvironment: Friends or Foes? IJMS 2019, 20, 1714.
- Liu, D.; You, M.; Xu, Y.; Li, F.; Zhang, D.; Li, X.; Hou, Y. Inhibition of Curcumin on Myeloid-Derived Suppressor Cells Is Requisite for Controlling Lung Cancer. Int. Immunopharmacol. 2016, 39, 265–272.
- Halder, R.C.; Almasi, A.; Sagong, B.; Leung, J.; Jewett, A.; Fiala, M. Curcuminoids and ω-3 Fatty Acids with Anti-Oxidants Potentiate Cytotoxicity of Natural Killer Cells against Pancreatic Ductal Adenocarcinoma Cells and Inhibit Interferon γ Production. Front. Physiol. 2015, 6, 129.
- Lee, Y.; Shin, H.; Kim, J. In Vivo Anti-Cancer Effects of Resveratrol Mediated by NK Cell Activation. J. Innate Immun. 2021, 13, 94–106.
- Zhang, X.; Cook, K.L.; Warri, A.; Cruz, I.M.; Rosim, M.; Riskin, J.; Helferich, W.; Doerge, D.; Clarke, R.; Hilakivi-Clarke, L. Lifetime Genistein Intake Increases the Response of Mammary Tumors to Tamoxifen in Rats. Clin. Cancer Res. 2017, 23, 814–824.
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025.
- Zhou, Z.; Liu, Y.; Song, W.; Jiang, X.; Deng, Z.; Xiong, W.; Shen, J. Metabolic Reprogramming Mediated PD-L1 Depression and Hypoxia Reversion to Reactivate Tumor Therapy. J. Control. Release 2022, 352, 793–812.
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452.
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol Targets PD-L1 Glycosylation and Dimerization to Enhance Antitumor T-Cell Immunity. Aging 2020, 12, 8–34.
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin Suppresses PD-L1 Expression in Melanoma and Host Dendritic Cells to Elicit Synergistic Therapeutic Effects. J. Exp. Clin. Cancer Res. 2018, 37, 261.
- Jiang, Z.-B.; Wang, W.-J.; Xu, C.; Xie, Y.-J.; Wang, X.-R.; Zhang, Y.-Z.; Huang, J.-M.; Huang, M.; Xie, C.; Liu, P.; et al. Luteolin and Its Derivative Apigenin Suppress the Inducible PD-L1 Expression to Improve Anti-Tumor Immunity in KRAS-Mutant Lung Cancer. Cancer Lett. 2021, 515, 36–48.
- Liao, F.; Liu, L.; Luo, E.; Hu, J. Curcumin Enhances Anti-Tumor Immune Response in Tongue Squamous Cell Carcinoma. Arch. Oral Biol. 2018, 92, 32–37.
- Guo, L.; Li, H.; Fan, T.; Ma, Y.; Wang, L. Synergistic Efficacy of Curcumin and Anti-Programmed Cell Death-1 in Hepatocellular Carcinoma. Life Sci. 2021, 279, 119359.
- Liu, K.; Sun, Q.; Liu, Q.; Li, H.; Zhang, W.; Sun, C. Focus on Immune Checkpoint PD-1/PD-L1 Pathway: New Advances of Polyphenol Phytochemicals in Tumor Immunotherapy. Biomed. Pharmacother. 2022, 154, 113618.
- Yang, M.; Li, Z.; Tao, J.; Hu, H.; Li, Z.; Zhang, Z.; Cheng, F.; Sun, Y.; Zhang, Y.; Yang, J.; et al. Resveratrol Induces PD-L1 Expression through Snail-Driven Activation of Wnt Pathway in Lung Cancer Cells. J. Cancer Res. Clin. Oncol. 2021, 147, 1101–1113.
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370.
- Mohos, V.; Fliszár-Nyúl, E.; Lemli, B.; Zsidó, B.Z.; Hetényi, C.; Mladěnka, P.; Horký, P.; Pour, M.; Poór, M. Testing the Pharmacokinetic Interactions of 24 Colonic Flavonoid Metabolites with Human Serum Albumin and Cytochrome P450 Enzymes. Biomolecules 2020, 10, 409.
- Fatima, A.; Khan, M.S.; Ahmad, M.W. Therapeutic Potential of Equol: A Comprehensive Review. Curr. Pharm. Des. 2020, 26, 5837–5843.
- Tuli, H.S.; Kumar, A.; Sak, K.; Aggarwal, D.; Gupta, D.S.; Kaur, G.; Vashishth, K.; Dhama, K.; Kaur, J.; Saini, A.K.; et al. Gut Microbiota-Assisted Synthesis, Cellular Interactions and Synergistic Perspectives of Equol as a Potent Anticancer Isoflavone. Pharmaceuticals 2022, 15, 1418.
- Rogovskii, V.S. The Therapeutic Potential of Urolithin A for Cancer Treatment AndPrevention. Curr. Cancer Drug Targets 2022, 22, 717–724.
- Al-Harbi, S.A.; Abdulrahman, A.O.; Zamzami, M.A.; Khan, M.I. Urolithins: The Gut Based Polyphenol Metabolites of Ellagitannins in Cancer Prevention, a Review. Front. Nutr. 2021, 8, 647582.
- Zhang, J.; Ren, L.; Yu, M.; Liu, X.; Ma, W.; Huang, L.; Li, X.; Ye, X. S-Equol Inhibits Proliferation and Promotes Apoptosis of Human Breast Cancer MCF-7 Cells via Regulating MiR-10a-5p and PI3K/AKT Pathway. Arch. Biochem. Biophys. 2019, 672, 108064.
- Zou, Y.; Wang, Y.; Cai, Y.; Ma, D. Effects of equol on proliferation of colorectal cancer HCT-15 cell. Wei Sheng Yan Jiu 2019, 48, 803–806.
- Magee, P.J.; Allsopp, P.; Samaletdin, A.; Rowland, I.R. Daidzein, R-(+)Equol and S-(-)Equol Inhibit the Invasion of MDA-MB-231 Breast Cancer Cells Potentially via the down-Regulation of Matrix Metalloproteinase-2. Eur. J. Nutr. 2014, 53, 345–350.
- Bao, C.; Namgung, H.; Lee, J.; Park, H.-C.; Ko, J.; Moon, H.; Ko, H.W.; Lee, H.J. Daidzein Suppresses Tumor Necrosis Factor-α Induced Migration and Invasion by Inhibiting Hedgehog/Gli1 Signaling in Human Breast Cancer Cells. J. Agric. Food Chem. 2014, 62, 3759–3767.
- Brown, N.M.; Belles, C.A.; Lindley, S.L.; Zimmer-Nechemias, L.D.; Zhao, X.; Witte, D.P.; Kim, M.O.; Setchell, K.D.R. The Chemopreventive Action of Equol Enantiomers in a Chemically Induced Animal Model of Breast Cancer. Carcinogenesis 2010, 31, 886–893.
- Yu, X.; Zou, Y.Q.; Wang, Y.; Chen, Z.K.; Ma, D.F. Equol and its enantiomers inhibited urethane-induced lung cancer in mice. Beijing Da Xue Xue Bao Yi Xue Ban 2022, 54, 244–248.
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; García-Villalba, R.; Tomás-Barberán, F.A.; Espín, J.C. Antiproliferative Activity of the Ellagic Acid-Derived Gut Microbiota Isourolithin A and Comparison with Its Urolithin A Isomer: The Role of Cell Metabolism. Eur. J. Nutr. 2017, 56, 831–841.
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomé-Carneiro, J.; Tomás-Barberán, F.A.; García-Conesa, M.T.; Espín, J.C. Comprehensive Characterization of the Effects of Ellagic Acid and Urolithins on Colorectal Cancer and Key-Associated Molecular Hallmarks: MicroRNA Cell Specific Induction of CDKN1A (P21) as a Common Mechanism Involved. Mol. Nutr. Food Res. 2016, 60, 701–716.
- Mohammed Saleem, Y.I.; Albassam, H.; Selim, M. Urolithin A Induces Prostate Cancer Cell Death in P53-Dependent and in P53-Independent Manner. Eur. J. Nutr. 2020, 59, 1607–1618.
- El-Wetidy, M.S.; Ahmad, R.; Rady, I.; Helal, H.; Rady, M.I.; Vaali-Mohammed, M.-A.; Al-Khayal, K.; Traiki, T.B.; Abdulla, M.-H. Urolithin A Induces Cell Cycle Arrest and Apoptosis by Inhibiting Bcl-2, Increasing P53-P21 Proteins and Reactive Oxygen Species Production in Colorectal Cancer Cells. Cell Stress Chaperones 2021, 26, 473–493.
- Liu, C.-F.; Li, X.-L.; Zhang, Z.-L.; Qiu, L.; Ding, S.-X.; Xue, J.-X.; Zhao, G.-P.; Li, J. Antiaging Effects of Urolithin A on Replicative Senescent Human Skin Fibroblasts. Rejuvenation Res. 2019, 22, 191–200.
- Djedjibegovic, J.; Marjanovic, A.; Panieri, E.; Saso, L. Ellagic Acid-Derived Urolithins as Modulators of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 5194508.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
996
Revisions:
3 times
(View History)
Update Date:
02 Aug 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No