Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giovanna Dashi | -- | 1794 | 2023-08-01 12:55:22 | | | |
| 2 | Fanny Huang | -100 word(s) | 1694 | 2023-08-02 03:16:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Salokas, K.; Dashi, G.; Varjosalo, M. The Impact of Oncofusions in Cancer Research. Encyclopedia. Available online: https://encyclopedia.pub/entry/47492 (accessed on 27 June 2026).
Salokas K, Dashi G, Varjosalo M. The Impact of Oncofusions in Cancer Research. Encyclopedia. Available at: https://encyclopedia.pub/entry/47492. Accessed June 27, 2026.
Salokas, Kari, Giovanna Dashi, Markku Varjosalo. "The Impact of Oncofusions in Cancer Research" Encyclopedia, https://encyclopedia.pub/entry/47492 (accessed June 27, 2026).
Salokas, K., Dashi, G., & Varjosalo, M. (2023, August 01). The Impact of Oncofusions in Cancer Research. In Encyclopedia. https://encyclopedia.pub/entry/47492
Salokas, Kari, et al. "The Impact of Oncofusions in Cancer Research." Encyclopedia. Web. 01 August, 2023.
Copy Citation
Oncofusions, or cancer-associated fusion mutations, are driving forces in cancer development. Advanced sequencing technologies have revolutionized their identification, opening new avenues in cancer research. Oncofusions manipulate cellular signaling pathways and show promise as targets for therapy and diagnostic markers.
cancer
fusion mutation
oncofusion
cancer treatment
1. Introduction
Cancer-associated gene fusions, also known as oncofusions (OFs), are hybrid genes formed when two previously independent genes become juxtaposed (Figure 1). The formation of these fusions can stem from structural rearrangements, transcription read-through of neighboring genes, or the trans- and cis-splicing of pre-mRNAs. Gene fusions with oncogenic properties are referred to as oncofusions, and often act as driver mutations in different cancers [1]. The identification of the first oncofusion signifies a landmark in the study of cancer [2]. Since then, developments in the applicable methods have enabled an accelerating pace of discovery of new oncofusions in cancer samples, with the pace skyrocketing after massively parallel sequencing (MPS) became viable. Oncofusions have been identified extensively in solid tumors, leukemias, and lymphomas, and in both singular studies [3] and systemic sequencing studies [4][5].
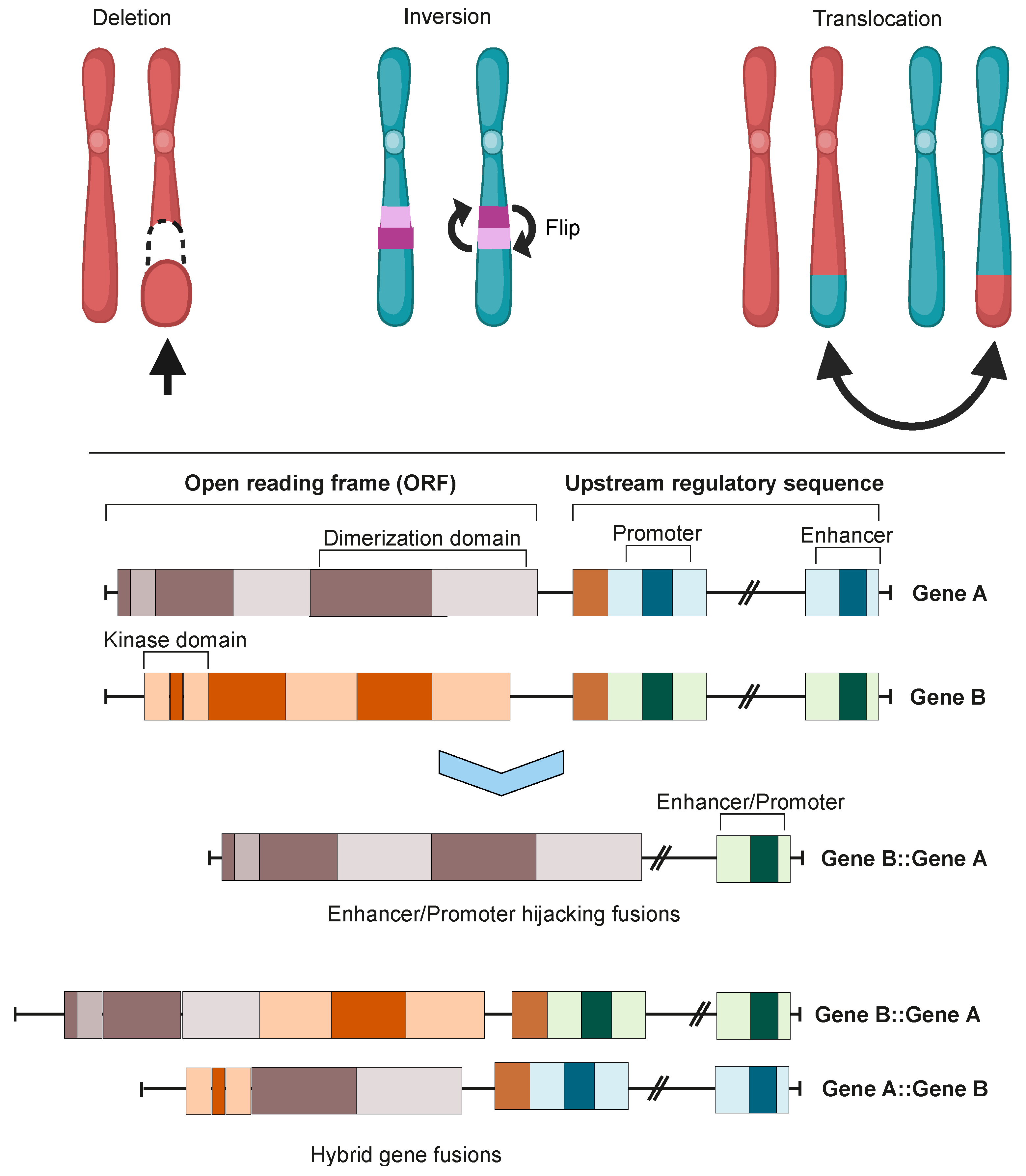
Figure 1. Chromosomal instability produces gene fusions. Top panel: chromosomal rearrangements leading to fusion mutations. Bottom panel: types of gene fusions. Depending on the specific fusion sites in both genes, the result can be either a hybrid gene fusion or a promoter- or enhancer-hijacking fusion. In both cases, the produced proteins can be either in-frame or out-of-frame. Images were created using BioRender.com.
Oncofusions drive cancer development primarily through two mechanisms: deregulation and the creation of hybrid genes. Deregulation occurs when one gene becomes linked to another’s regulatory region, typically resulting in the overexpression of an otherwise normal gene. The first conclusive evidence for the deregulation mechanism was provided by analyses of Burkitt lymphoma (BL) and chronic myeloid leukemia (CML). These oncofusions link the coding region of the MYC oncogene to the regulatory region of immunoglobulin, leading to MYC overexpression and subsequently oncogenesis [6][7][8]. The second oncogenic mechanism involves the fusion of the coding regions of two different genes to generate a hybrid gene, resulting in a functional chimeric protein such as BCR::ABL [2]. This oncofusion results in a fully functional protein with aberrant kinase activity, and is the best-described model example of abnormal protein function resulting from an oncofusion [9][10][11][12][13][14]. Indeed, oncofusions are known to disturb multiple critical cellular signaling pathways regulating proliferation, differentiation, and survival (Figure 2).
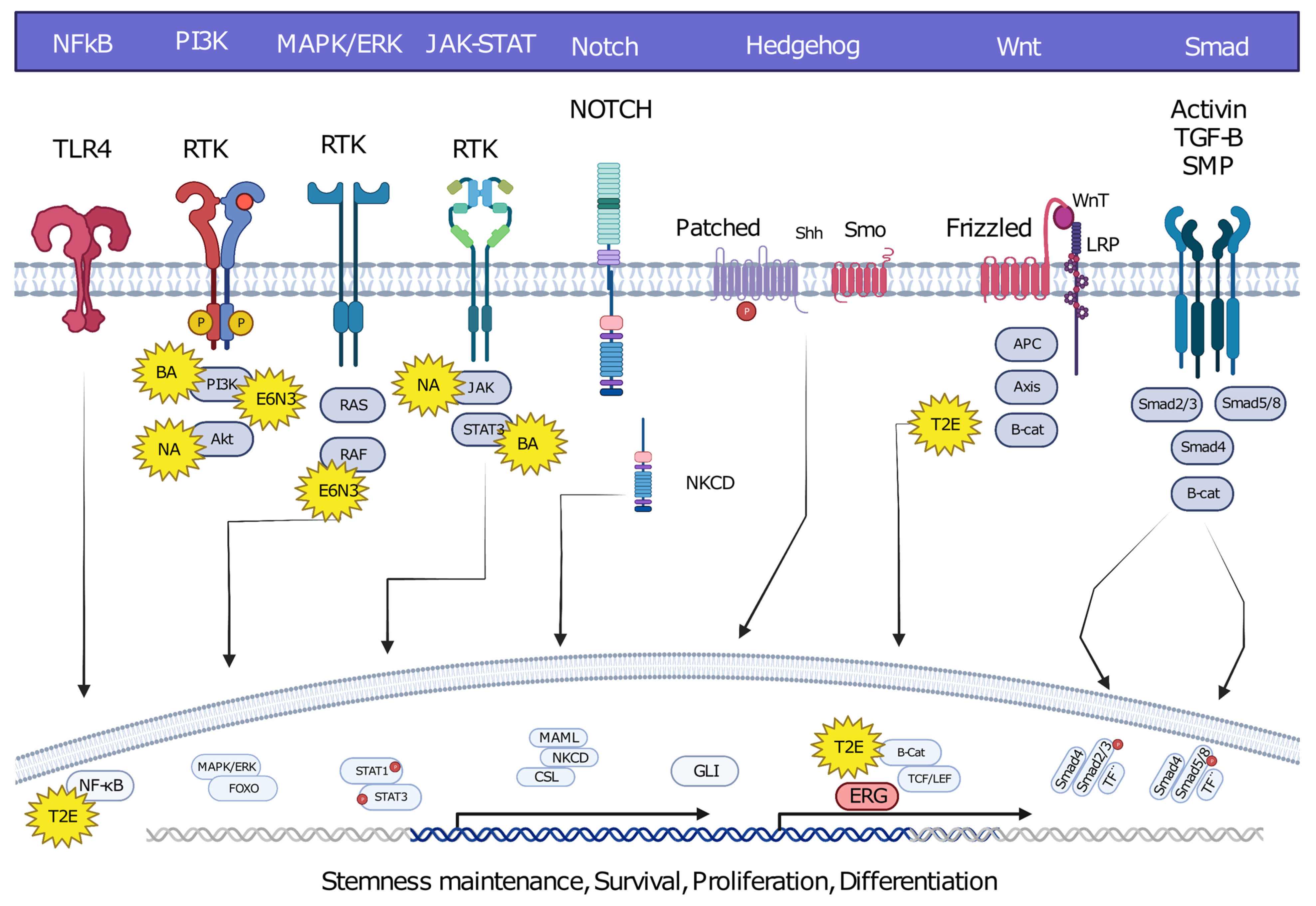
Figure 2. Hybrid oncofusions disrupt many receptor-dependent signaling pathways, whether downstream or upstream. Common fusion-affected pathways are highlighted along with well-known fusions affecting the corresponding pathways. BA: BCR::ABL, E6N3: ETV6::NTRK3, NA: NPM::ALK, T2E: TMPRESS2::ERG. Image was created using BioRender.com.
While oncofusions are typically somatic mutations, a few hereditary exceptions have been described [15]. Guided genomic methods such as quantitative real-time PCR (qPCR) and fluorescent in situ hybridization (FISH), together with extensive genomic research efforts like the Cancer Genome Atlas Project (TCGA) and the more recent Japanese Cancer Genome Atlas (JCGA), have contributed significantly to the identification of tens of thousands of oncofusions since the 1960s and 1970s [3][4][5]. The TCGA project has been particularly influential in shedding light on the scale, prevalence, and impact of oncofusions. The project propelled forward the development of both identification strategies and treatment options by molecularly characterizing over 20,000 cancer samples from 33 different cancer types.
The combined knowledge of all known oncofusions supports the significance of deregulation and hybrid gene formation in fusion oncogenicity, although other mechanisms may also play a role. Moreover, some oncofusions can drive cancer development via the inactivation of cancer suppressor genes, for example by coupling them to regulatory elements of genes that are not usually expressed [16][17]. The major challenge with vast numbers of possible, probable, and confirmed oncofusions is validation and relevance: many could be functionally irrelevant or barely expressed passenger mutations, and most have been described as random mutations arising from chance events [18]. However, as several studies highlight, non-driver mutations can still be very valuable for cancer treatment due to their role in producing neoantigens [19][20].
2. Oncofusion Formation
Integration of transcriptomic and genomic data has estimated that approximately two thirds of fusion mutations stem from erroneous repair of DNA DSBs [18]. Erroneous DNA DSB repair can lead to genome instability, inducing mutagenesis and genomic rearrangements such as deletions, inversions, duplications, and translocations. All of these genomic rearrangements can result in the formation of oncofusions [21].
Although the majority of fusions transpire in “gene-rich” areas of the genome [18], intragenic fusions have been assessed to constitute only 38% of fusion events [22]. These involve the joining of one gene’s coding sequence with the coding sequence of another, resulting in an in-frame gene fusion such as BCR::ABL1 or TMPRSS::ERG. A large proportion of oncofusions have intergenic regions from at least one fusion partner (Figure 3A), but can sometimes still produce proteins where the 3′ fusion partner is also in-frame [22][23]. Some intergenic oncofusions are the result of a gene with an otherwise weak promoter fusing to a stronger promoter or enhancer (e.g., IGH-MYC in BL) [24]. These events are known as promoter or enhancer swapping [22].

Figure 3. General assessment of fusions from tumorfusions.org database. (A) Count of fusions based on frame prediction. (B) Fusion distribution based on gene type. N/A denotes genes which could not be categorized definitively as protein kinases, transcription factors, tumor suppressors, or other oncogenes. (C) Distribution of fusions based on frame prediction and gene type. Only fusions that could be assigned to one of the four gene groups are included.
For the purpose of a general assessment of fusions and participating genes, researchers downloaded data from tumorfusions.org. Although tumorfusions.org data only reflect data from TCGA and different analyses can produce different results, these data were deemed sufficiently accurate for an overall representation of oncofusions. Researchers conducted an analysis of data downloaded from tumorfusions.org, focusing on key genes participating in fusions. This dataset was then cross-verified and annotated with information from the COSMIC, ChimerDB, and Uniprot databases. The genes were subsequently categorized into four main groups for an in-depth analysis: kinases, transcription factors, non-kinase or TF tumor suppressors, and other oncogenes. The majority of genes participating in fusions, such as VMP1 and TACC3, do not belong to any of these groups (Figure 3B). Of the annotated genes, in-frame fusions, kinases, and transcription factors are equally common, and thus it is no surprise that much research has focused on them in recent years (Figure 3C).
On the kinase side, the majority of the in-frame fusions originated from combinations of FGFR3, BRAF, RET, NF1, and FGFR2, with 36, 33, 28, 18, and 18 fusions, respectively, with a variety of partners (FGFR3 combined in particular with TACC3) (Figure 4A). A group that has often been identified as a major contributor to fusions, as well as cancers driven by other mutations, is the RTKs, especially the FGFR and NTRK subfamilies, as well as RET and ALK.
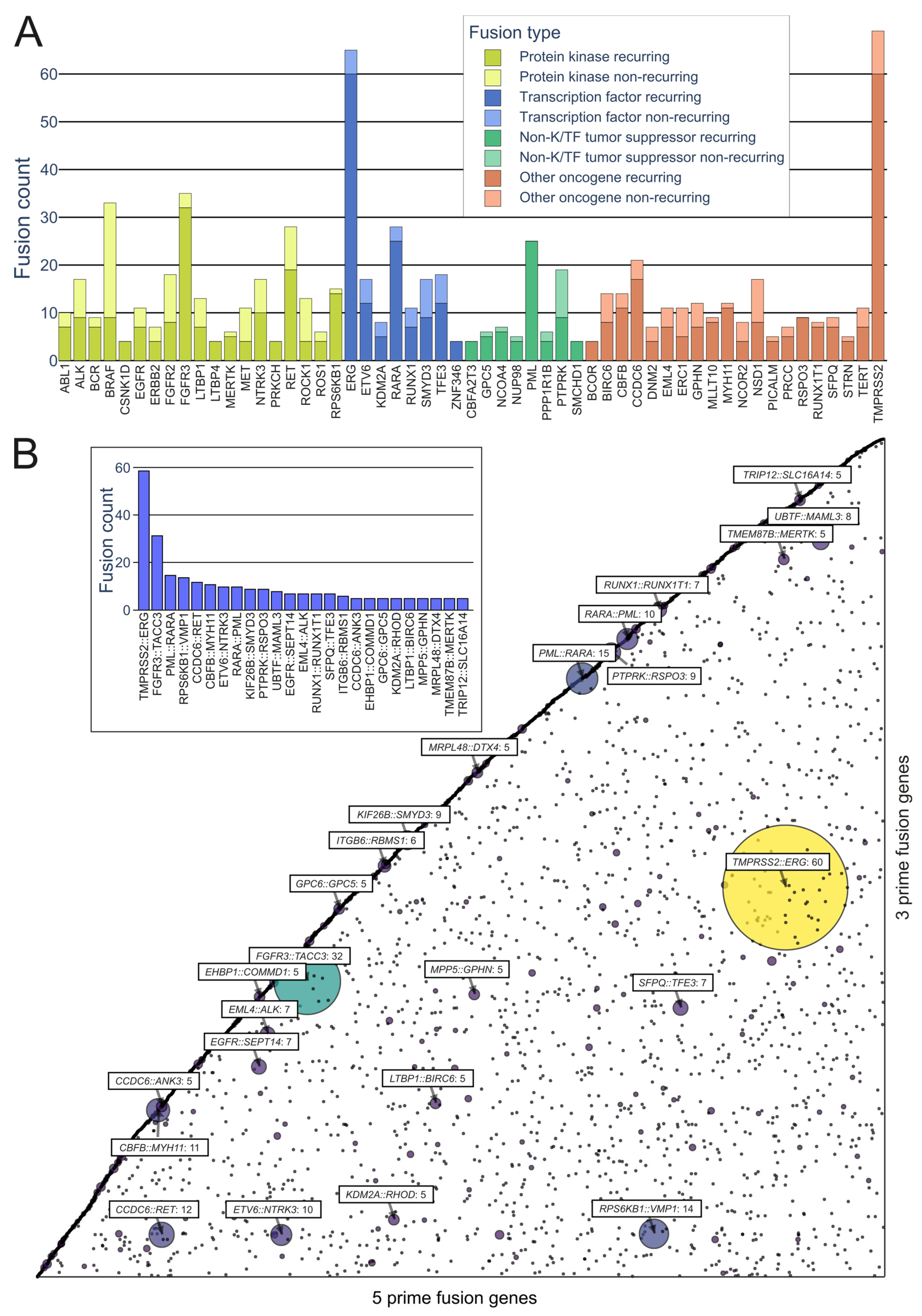
Figure 4. Representation of recurring fusions from tumorfusions.org data. (A) Count of recurring and non-recurring fusions per gene in each gene type category. Included are genes present in at least 5 recurring fusions. (B) Scatter plot of all in-frame fusions. 5 prime genes are on the x axis and 3 prime genes are on the y axis. Size and color of the bubbles correspond to the number of fusions in the tumorfusions.org dataset. Gene pairs with 5 or more recurring fusions identified are highlighted. Inset: highlighted fusions in bar plot format.
While kinases hold significant attention in oncofusion research, transcription factors also play a considerable role. For instance, the most common genes involved in oncofusions were ERG, RARA, TFE3, and ETV6, with 65, 28, 18, and 17 fusions, respectively. The group in general was driven by the TMPRSS2::ERG fusion, which was overall the most common fusion found, with 60 in-frame fusions identified in the tumorfusions.org TCGA dataset.
Genomic rearrangements are present in roughly half of hematopoietic cancers as well as up to 90% of solid tumors [25], many of which harbor oncofusions [26]. The products of these oncofusions play pivotal roles in tumor evolution and progression [27][28].
3. Oncofusions’ Role in Cancer Development
Cancer development has historically been viewed as a slow accumulation of mutations which increases genomic instability, ultimately resulting in both driver and passenger mutations [29]. Instability takes place at the nucleotide level, causing small-scale changes such as point mutations and minor deletions, as well as at the chromosomal level, prompting more substantial rearrangements of entire chromosomal segments [30].
Following the first large-scale identification of oncofusions in MPS studies, it was estimated in 2007 that fusions accounted for roughly 20% of cancer morbidity and represented early events in cancer genesis [28]. More recently, during the JCGA project, known oncofusions from a limited panel of 491 were identified as driver mutations in 12.9% of cancer specimens [5]. Identification varies by cancer type. It is noteworthy that the prevalence of fusions is significant in specific cancers—they are identified in over 90% of all lymphomas [31] and more than 30% of soft-tissue tumors [32]. Other research has suggested that fusions drive 16.5% of human cancers and act as solitary drivers in 1% [33]. These estimates are likely to be reasonably accurate.
Prominent Oncofusions in Cancer
Various kinase and transcription factor fusions are frequently emphasized as driver oncofusions [34][35][36][37][38][39][40][41][42]. In the case of transcription factor fusions, prevalent mechanisms of cancer formation revolve around the DNA-binding domain. Kinase oncofusions often lead to the loss of kinase domain regulation and the acquisition of an alternative dimerization mechanism, such as the coiled-coil domain of TACC3 in the FGFR3::TACC3 oncofusion [43].
In addition to gene groups, specific oncofusions have been highlighted as crucial early steps in the initiation of tumorigenesis, or as crucial contributors to tumor morbidity [44][45][46][47]. Furthermore, many oncogenes seem to require the occurrence of specific fusion events to unleash their oncogenic potential. In such instances, there is minimal variability in how the fusion occurs, and they are frequently recognized as recurrent oncofusions in large-scale studies. For example, the oncogenic potential of the IGH::BCL2 oncofusion is derived from the combination of the anti-apoptotic BCL2 with the highly expressed immunoglobulin locus of IGH [48][49]. A similar mechanism is often present in prostate cancer, where the TMPRSS2::ERG oncofusion is found in roughly 50% of tumors and MAN2A1::FER, MTOR::TP52BP1, and SLC45A2::AMACR occur at lower frequencies. The common factor uniting these fusions is the association of an oncogene with a more active promoter region [50][51][52][53].
References
- Latysheva, N.S.; Babu, M.M. Discovering and Understanding Oncogenic Gene Fusions through Data Intensive Computational Approaches. Nucleic Acids Res. 2016, 44, 4487–4503.
- Nowell, P.; Hungerford, D. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947.
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120.
- Nagashima, T.; Yamaguchi, K.; Urakami, K.; Shimoda, Y.; Ohnami, S.; Ohshima, K.; Tanabe, T.; Naruoka, A.; Kamada, F.; Serizawa, M.; et al. Japanese Version of The Cancer Genome Atlas, JCGA, Established Using Fresh Frozen Tumors Obtained from 5143 Cancer Patients. Cancer Sci. 2020, 111, 687–699.
- Erikson, J.; Nishikura, K.; ar-Rushdi, A.; Finan, J.; Emanuel, B.; Lenoir, G.; Nowell, P.C.; Croce, C.M. Translocation of an Immunoglobulin Kappa Locus to a Region 3′ of an Unrearranged c-Myc Oncogene Enhances c-Myc Transcription. Proc. Natl. Acad. Sci. USA 1983, 80, 7581–7585.
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human C-Myc Onc Gene Is Located on the Region of Chromosome 8 That Is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827.
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the C-Myc Gene into the Immunoglobulin Heavy Chain Locus in Human Burkitt Lymphoma and Murine Plasmacytoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841.
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural Organization of the Bcr Gene and Its Role in the Ph’ Translocation. Nature 1985, 315, 758–761.
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused Transcript of Abl and Bcr Genes in Chronic Myelogenous Leukaemia. Nature 1985, 315, 550–554.
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The Chronic Myelogenous Leukemia-Specific P210 Protein Is the Product of the Bcr/Abl Hybrid Gene. Science 1986, 233, 212–214.
- Fainstein, E.; Marcelle, C.; Rosner, A.; Canaani, E.; Gale, R.P.; Dreazen, O.; Smith, S.D.; Croce, C.M. A New Fused Transcript in Philadelphia Chromosome Positive Acute Lymphocytic Leukaemia. Nature 1987, 330, 386–388.
- Reckel, S.; Hamelin, R.; Georgeon, S.; Armand, F.; Jolliet, Q.; Chiappe, D.; Moniatte, M.; Hantschel, O. Differential Signaling Networks of Bcr-Abl P210 and P190 Kinases in Leukemia Cells Defined by Functional Proteomics. Leukemia 2017, 31, 1502–1512.
- Cutler, J.A.; Tahir, R.; Sreenivasamurthy, S.K.; Mitchell, C.; Renuse, S.; Nirujogi, R.S.; Patil, A.H.; Heydarian, M.; Wong, X.; Wu, X.; et al. Differential Signaling through P190 and P210 BCR-ABL Fusion Proteins Revealed by Interactome and Phosphoproteome Analysis. Leukemia 2017, 31, 1513–1524.
- Efremov, G.D. Hemoglobins Lepore and Anti-Lepore. Hemoglobin 1978, 2, 197–233.
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The Emerging Complexity of Gene Fusions in Cancer. Nat. Rev. Cancer 2015, 15, 371–381.
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of Gene Fusions in Epithelial Cancers: Seq and Ye Shall Find. Genome Med. 2015, 7, 129.
- Johansson, B.; Mertens, F.; Schyman, T.; Björk, J.; Mandahl, N.; Mitelman, F. Most Gene Fusions in Cancer Are Stochastic Events. Genes Chromosomes Cancer 2019, 58, 607–611.
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-Neo: A Pipeline for Personalized Gene Fusion Neoantigen Discovery. Bioinformatics 2017, 33, 555–557.
- Yi-Mi, W.; Cieslik, M.; Lonigro, R.J.; Pankaj, V.; Reimers, M.A.; Xuhong, C.; Yu, N.; Lisha, W.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14.
- So, A.; Le Guen, T.; Lopez, B.S.; Guirouilh-Barbat, J. Genomic Rearrangements Induced by Unscheduled DNA Double Strand Breaks in Somatic Mammalian Cells. FEBS J. 2017, 284, 2324–2344.
- Yun, J.W.; Yang, L.; Park, H.-Y.; Lee, C.-W.; Cha, H.; Shin, H.-T.; Noh, K.-W.; Choi, Y.-L.; Park, W.-Y.; Park, P.J. Dysregulation of Cancer Genes by Recurrent Intergenic Fusions. Genome Biol. 2020, 21, 166.
- Namba, S.; Ueno, T.; Kojima, S.; Kobayashi, K.; Kawase, K.; Tanaka, Y.; Inoue, S.; Kishigami, F.; Kawashima, S.; Maeda, N.; et al. Transcript-Targeted Analysis Reveals Isoform Alterations and Double-Hop Fusions in Breast Cancer. Commun. Biol. 2021, 4, 1320.
- Ferrad, M.; Ghazzaui, N.; Issaoui, H.; Cook-Moreau, J.; Denizot, Y. Mouse Models of C-Myc Deregulation Driven by IgH Locus Enhancers as Models of B-Cell Lymphomagenesis. Front. Immunol. 2020, 11, 1564.
- Gagos, S.; Irminger-Finger, I. Chromosome Instability in Neoplasia: Chaotic Roots to Continuous Growth. Int. J. Biochem. Cell Biol. 2005, 37, 1014–1033.
- Takeuchi, K. Discovery Stories of RET Fusions in Lung Cancer: A Mini-Review. Front. Physiol. 2019, 10, 216.
- Long, M.; Langley, C.H. Natural Selection and the Origin of Jingwei, a Chimeric Processed Functional Gene in Drosophila. Science 1993, 260, 91–95.
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Lee, J.-K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. 2016, 11, 283–312.
- Lobato, M.N.; Metzler, M.; Drynan, L.; Forster, A.; Pannell, R.; Rabbitts, T.H. Modeling Chromosomal Translocations Using Conditional Alleles to Recapitulate Initiating Events in Human Leukemias. J. Natl. Cancer Inst. Monogr. 2008, 2008, 58–63.
- Mertens, F.; Antonescu, C.R.; Mitelman, F. Gene Fusions in Soft Tissue Tumors: Recurrent and Overlapping Pathogenetic Themes. Genes Chromosomes Cancer 2016, 55, 291–310.
- Gao, Q.; Liang, W.-W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.-W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3.
- Salokas, K.; Weldatsadik, R.G.; Varjosalo, M. Human Transcription Factor and Protein Kinase Gene Fusions in Human Cancer. Sci. Rep. 2020, 10, 14169.
- Nagy, Z.; Jeselsohn, R. ESR1 Fusions and Therapeutic Resistance in Metastatic Breast Cancer. Front. Oncol. 2022, 12, 1037531.
- Yu, L.; Davis, I.J.; Liu, P. Regulation of EWSR1-FLI1 Function by Post-Transcriptional and Post-Translational Modifications. Cancers 2023, 15, 382.
- Apfelbaum, A.A.; Wrenn, E.D.; Lawlor, E.R. The Importance of Fusion Protein Activity in Ewing Sarcoma and the Cell Intrinsic and Extrinsic Factors That Regulate It: A Review. Front. Oncol. 2022, 12, 1044707.
- Bowling, G.C.; Rands, M.G.; Dobi, A.; Eldhose, B. Emerging Developments in ETS-Positive Prostate Cancer Therapy. Mol. Cancer Ther. 2023, 22, 168–178.
- Shen, Z.; Qiu, B.; Li, L.; Yang, B.; Li, G. Targeted Therapy of RET Fusion-Positive Non-Small Cell Lung Cancer. Front. Oncol. 2022, 12, 1033484.
- Sorokin, M.; Rabushko, E.; Rozenberg, J.M.; Mohammad, T.; Seryakov, A.; Sekacheva, M.; Buzdin, A. Clinically Relevant Fusion Oncogenes: Detection and Practical Implications. Ther. Adv. Med. Oncol. 2022, 14, 17588359221144108.
- Hagstrom, M.; Fumero-Velázquez, M.; Dhillon, S.; Olivares, S.; Gerami, P. An Update on Genomic Aberrations in Spitz Naevi and Tumours. Pathology 2023, 55, 196–205.
- Chu, Y.-H. This Is Your Thyroid on Drugs: Targetable Mutations and Fusions in Thyroid Carcinoma. Surg. Pathol. Clin. 2023, 16, 57–73.
- Wang, C.G.; Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Donoghue, D.J. Oncogenic Driver FGFR3-TACC3 Requires Five Coiled-Coil Heptads for Activation and Disulfide Bond Formation for Stability. Oncotarget 2023, 14, 133–145.
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.-C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E.; et al. DNAJB1-PRKACA Fusion Kinase Interacts with β-Catenin and the Liver Regenerative Response to Drive Fibrolamellar Hepatocellular Carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 13076–13084.
- Matissek, K.J.; Onozato, M.L.; Sun, S.; Zheng, Z.; Schultz, A.; Lee, J.; Patel, K.; Jerevall, P.-L.; Saladi, S.V.; Macleay, A.; et al. Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor-Positive Breast Cancer. Cancer Discov. 2018, 8, 336–353.
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep. 2018, 24, 1434–1444.e7.
- Kim, J.; Kim, S.; Ko, S.; In, Y.; Moon, H.-G.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.-H.; Ju, Y.S.; et al. Recurrent Fusion Transcripts Detected by Whole-Transcriptome Sequencing of 120 Primary Breast Cancer Samples. Genes Chromosomes Cancer 2015, 54, 681–691.
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099.
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442.
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648.
- Yu, Y.-P.; Liu, P.; Nelson, J.; Hamilton, R.L.; Bhargava, R.; Michalopoulos, G.; Chen, Q.; Zhang, J.; Ma, D.; Pennathur, A.; et al. Identification of Recurrent Fusion Genes across Multiple Cancer Types. Sci. Rep. 2019, 9, 1074.
- Yu, Y.P.; Ding, Y.; Chen, Z.; Liu, S.; Michalopoulos, A.; Chen, R.; Gulzar, Z.G.; Yang, B.; Cieply, K.M.; Luvison, A.; et al. Novel Fusion Transcripts Associate with Progressive Prostate Cancer. Am. J. Pathol. 2014, 184, 2840–2849.
- Yu, J.; Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, G.X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
951
Revisions:
2 times
(View History)
Update Date:
02 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No