Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuseok Moon | -- | 2460 | 2023-07-31 03:41:56 | | | |
| 2 | Catherine Yang | Meta information modification | 2460 | 2023-07-31 03:45:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moon, Y. Mucosal Etiologies of Renal Injuries Caused by PM. Encyclopedia. Available online: https://encyclopedia.pub/entry/47412 (accessed on 27 July 2026).
Moon Y. Mucosal Etiologies of Renal Injuries Caused by PM. Encyclopedia. Available at: https://encyclopedia.pub/entry/47412. Accessed July 27, 2026.
Moon, Yuseok. "Mucosal Etiologies of Renal Injuries Caused by PM" Encyclopedia, https://encyclopedia.pub/entry/47412 (accessed July 27, 2026).
Moon, Y. (2023, July 31). Mucosal Etiologies of Renal Injuries Caused by PM. In Encyclopedia. https://encyclopedia.pub/entry/47412
Moon, Yuseok. "Mucosal Etiologies of Renal Injuries Caused by PM." Encyclopedia. Web. 31 July, 2023.
Copy Citation
Particulate matter (PM) is a microscopic aerosol mixture of solid particles and liquid droplets found in the air, derived from natural or anthropogenic origins, including the combustion of fossil fuels and industrial effluents. Among the early responsive tissues to PM exposure, the mucosal barrier of the airway and alimentary tract may be a crucial source of pathologic mediators leading to inflammatory renal diseases, including chronic kidney disease (CKD).
particulate matter
mucosal exposure
microbiota
chronic kidney disease
1. Mucosal Exposure and Translocation of PM
The airway is the primary route of exposure to PM [1]. The penetration of particles is associated with their size, shape, and chemical composition. Generally, PM10 can penetrate the deepest parts of the lungs, such as the bronchioles or alveoli. Average exposure to PM10 is associated with non-accidental mortality in patients with chronic obstructive pulmonary disease (COPD), especially those diagnosed with asthma-COPD overlap [2]. Moreover, the adverse effects of PM10 exposure are relatively severe in women and nonsmokers [2]. Fine particulate matter (PM2.5) reaches the gas exchange regions of the lung alveoli, and only nanoscale particles can pass through the air-blood barrier in the acinar region through endocytosis or diffusion and affect other organs via the circulation [3][4][5]. However, larger particles, such as PM10 can deposit or be internalized by macrophages, causing detrimental effects on the local tissues and neighboring barrier. Disruption of the barrier would allow translocation of more PM to the circulation and increase the risk to the extrapulmonary organs.
Particles in inhaled air are cleared by a series of filtration systems. These particles become entrapped in the mucosal layer, and mucociliary transport quickly clears the inhaled PM from the lungs. In the airway regions lacking mucus transport via cilia movement, alveolar macrophages play a crucial role in the defense by phagocytosing foreign particles [6]. The biokinetic fate of inhaled ultrafine radiolabeled particles was examined in rodents [4]. The clearance of an overwhelming proportion of the particles, including ultrafine and micron-sized particles, is mostly mediated by macrophages that transport particles from the peripheral lungs to the larynx, with subsequent passage through the gut and fecal excretion [4]. In addition to gastrointestinal translocation from the airway, PM can enter water and food supply systems directly, and ultimately reach the gastrointestinal tract in humans [7][8]. Each individual ingests approximately 1012–1014 particles per day (based on a typical Western diet) and the gut mucosa can absorb 1% of ingested PM (109–1012 particles per day) given the huge exposure surface of the gut mucosa. In addition to lung exposure, the gastrointestinal tract is another primary deposition site of PM that displays potent early stress responses affecting disease outcomes.
2. Effects of PM on the Mucosal Barrier
Extensive parts of the airway and gut linings secrete viscous gel-like substances, known as mucins, as the first line of defense. They are large, highly glycosylated molecules that interact with exogenous substances; this interaction is a critical regulatory step in the migration of particulates to the underlying epithelia through the mucin mesh. Components of PM can directly disrupt this mucosal barrier by lowering the functional efficiency of the mucin structural network. Mechanistically, the collapse phenomenon in the arrangement of mucin networks is speculated to be due to chemical or physical interactions between mucins and external particulate components [9]. The dense connectivity of mucin fiber network loosens as PM-binding increases, with openings for luminal matter, including microbiota, nutrients, and xenobiotic agents. In addition to the impact on the mucosal barrier, PM ingestion can disrupt epithelial permeability [1][10][11]. Permeated PM generates reactive oxygen species (ROS) in epithelial cells [12], which decreases barrier integrity by rearranging or interrupting the epithelial junction in the epithelial lining [13][14]. In vitro evaluation showed reduced transepithelial resistance of the monolayers and structural changes in the tight junctions [1]. The deposition of PM correlated with diminished transcription levels of the tight junction protein 1 and occludin, and with histological evidence of modifications in tight junction organization [1]. Moreover, ingested PM causes gut permeability and aggravates colonic inflammation owing to alterations in cytokine networks [15][16]. In addition to the junctional disruption of the epithelial barrier, PM can cause epithelial cell apoptosis via ROS generation from the mitochondria induced by nuclear transcription factor NF-κB activation [1]. Among the various chemical components of PM, the oxidation products of primary gases are closely associated with a reduction in lifespan expectancy [17]. Conversely, a reduction in sulfate or ammonium is associated with an increase in life expectancy. To examine gene regulation, gene expression profiles were analyzed in cells exposed to PM components, such as sulfate, nitrate, and ammonium as representative life-threatening oxidation products of the primary gases, and endotoxins as a representative microbial product (Figure 1).
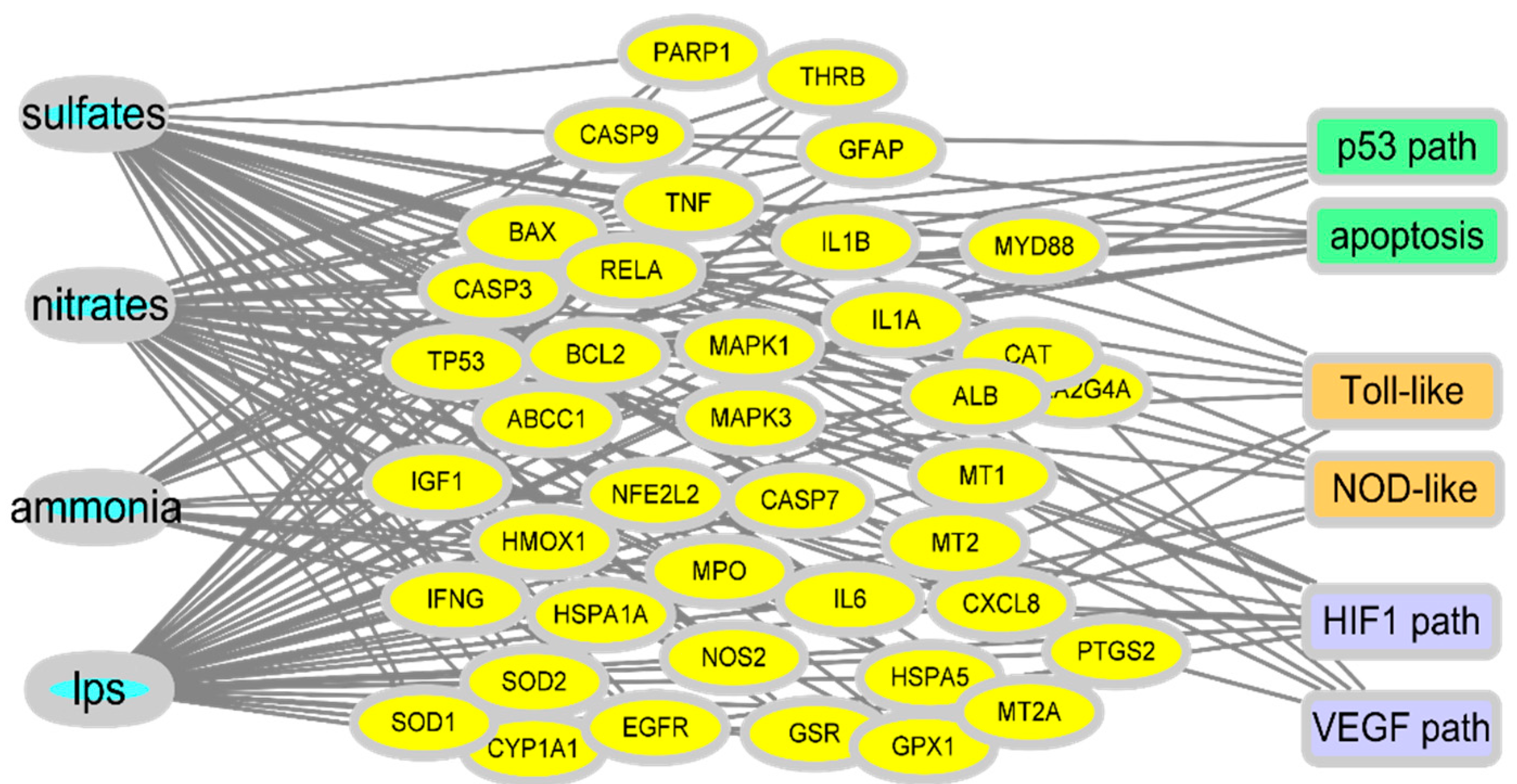
Figure 1. Prediction of gene network in PM-exposed mucosa. Gene expression profiles in the comparative toxicogenomic database in response to oxidation products of the primary gases and endotoxins as a representative microbial product. Based on PM exposure-linked gene sets, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to predict events in the insulted tissues.
In response to exposure to the PM-derived mixture, a network of target genes was revealed. As oxidative radical stress is considered to be the etiology of PM-induced tissue injury, many key network genes are involved in the cell death pathway, including p53. Moreover, another key feature of the network was the association of exposure to inflammatory stress signaling (toll-like receptor and NOD-like receptors) and hypoxia (HIF1 and VEGF pathway). All the molecular associations with cell death, inflammation, and hypoxia signaling pathways indicate the pathological outcomes in the exposed mucosal barrier during PM exposure. Furthermore, the disruption of gut permeability and epithelial injuries is subsequently associated with increased microbial access to the underlying mucosal immune tissues and cells, leading to inflammatory responses and changes in the mucosal microbial community.
3. Impact of PM on the Microbial Community
The microbiota harbors a complex and dynamic population of microbes, including bacteria, fungi, protozoa, and viruses, which form a continuous microbial community [18] that is built over a lifetime and plays crucial roles in metabolism and immunity in humans and animals [19]. Microbiota are responsive to changes in the luminal environment, such as nutrients and xenobiotic agents. Exposure to PM is thus a key cause of bacterial community changes, which impact immunity and other host physiology. In particular, PM components can induce oxidative stress in gut microbes, leading to the collapse of their community and inducing unexpected health risks in hosts, especially in people with chronic underlying disorders [20]. Long-term exposure to PM2.5 may contribute to increased risks of metabolic disorders, including T2D, in humans [21]. Exposure to PM2.5 was negatively associated with the alpha diversity index of the gut microbiota, and a lower diversity of the gut microbiota was associated with a higher risk of T2D [21]. In terms of richness, the composition of Firmicutes, Proteobacteria, or Verrucomicrobia phyla was negatively associated with both PM concentrations and the risk of T2D. Moreover, short-term exposure to PM resulted in a dose-dependent reduction in alpha diversity indices of microbiota in the nasal tract, an early mucosal exposure site in humans [22] although exposure to biomass fuel or motor vehicle exhaust elevated the abundance and alpha diversity of the lung microbiota in a rat model of exposure [23]. Depending on the exposure regimen or host species, different alterations of community patterns occur; these are a crucial factor in understanding the underlying pathogenesis of related diseases. Exposure to UFPs elevated cholesterol levels and reduced coprostanol levels in the cecum of mice [24]. Moreover, atherogenic lysophosphatidylcholine (18:1) and lysophosphatidic acid were found in the intestine and plasma of mice exposed to UFP. All these atherogenic lipids, including cholesterol, are potent mediators of pro-inflammatory responses, such as the recruitment of macrophages and neutrophils in the mucosa barrier. These lipids were negatively correlated with Actinobacteria, which was decreased by UFP exposure in a murine model [24]. Human epidemiological assessment in overweight and obese adolescents exposed to traffic-related air pollution also supported the negative correlation between changes in the microbiota and metabolic disorders [25]. Overall, the evidence in mice and humans provided a crucial insight into the contribution of PM-altered microbial communities to inflammatory cardiovascular diseases such as atherosclerosis. Moreover, PM-induced alteration of microbial communities can contribute to the metabolic activation of xenobiotic agents in PM [26]. In addition to effects on the community composition, PM-exposed gut microbiota displayed altered metabolic activities, which may affect the metabolism of endogenous biomolecules or the toxicity of PM-derived chemical components during mucosal exposure. Polycyclic aromatic hydrocarbons (PAHs) are among the most widespread organic pollutants generated by the incomplete combustion of fossil fuels and biomass. Although the parent PAH molecules are not estrogenic, in vitro evaluation in adult gut microbiota demonstrated the potent conversion of the parent PAHs to estrogenic hydroxyl metabolites, such as 1-OH pyrene and 7-OH benzo(a)pyrene [27]. In contrast, microbiota can protect against mutagen formation. For example, 2-nitrofluorene (NF), a representative nitro-PAH present in urban-air PM and diesel fuel emissions, can be reduced to 2-aminofluorene by the intestinal bacteria and is further acetylated to hydroxylated 2-acetylaminofluorene in the rat liver [28]. An alternate rat metabolism of NF results in the formation of mutagenic hydroxylated NF. However, mouse microflora tends to increase in 2-acetylaminofluorene, another DNA adduct from NF [28][29]. Therefore, depending on the host microbiota profile and the bioavailability of host cell metabolic enzymes, PM metabolites may be converted to either detrimental or inactive metabolites. Moreover, the PM-mediated alteration of the microbial community can determine the fate of PM-derived xenobiotics in human health and disease (Figure 2).
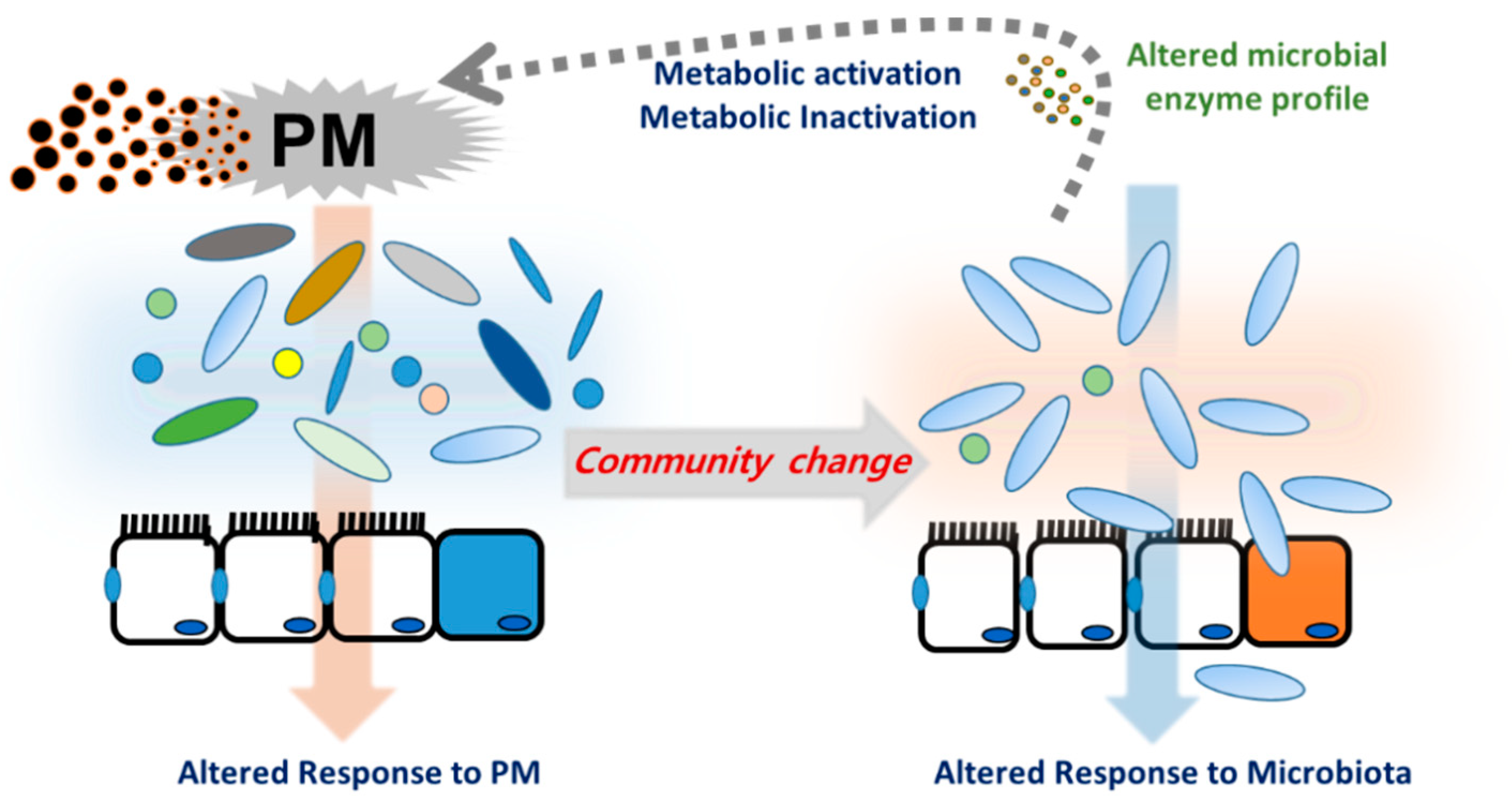
Figure 2. Particulate matter (PM)-induced alternation of mucosal microbial community. Host- and microbiota-derived xenobiotic metabolic enzymes are involved in metabolism of PM constituents. Moreover, PM can directly alter the microbial community, which inversely determines the fate of PM-derived xenobiotics or affect host responses during PM exposure.
4. Microbiota-Derived Uremic Solutes in Response to PM
The term uremic retention solutes (URS) refers to the components that accumulate in the blood and tissues during renal disease. Changes in the microbiota composition and community structure (dysbiosis) are associated with the production of 11 microbiota-derived uremic solutes [30][31]. Microbiota in normal conditions or dysbiosis can produce p-cresyl sulfate (PCS) from tyrosine, indoxyl sulfate (IS) from tryptophan, trimethylamine N-oxide from L-carnitine, dimethylglycine from choline, and glutarate from lysine [32]. Phenyl sulfate, cholate, hippurate, γ-guanidinobutyrate, 2-hydroxypentanoate, and phenaceturate are also considered URS. Moreover, the active metabolites in URS are formed by the combined actions of the microbial transformation and the host metabolic enzymes. For example, bacterial tryptophanase metabolizes dietary tryptophan to indole, which is subsequently hydroxylated to indoxyl by cytochrome P450 (CYP) isozyme 2E1 and finally sulfonated to indoxyl sulfate by sulfotransferases including SULT1A1 in the liver [33]. Therefore, hepatic dysfunction in patients with CKD and cirrhosis retards the formation of indoxyl sulfate and p-cresol sulfate due to impaired hepatic metabolism [34]. URS-induced renal injuries mostly result from inflammatory responses and radical production. Dysbiosis-induced indoxyl sulfate acts on the basolateral membrane of renal proximal tubular cells via binding to the organic anion transporter and causes inflammation and nephrotoxicity. PCS accumulates in kidney tubular cells, leading to the generation of ROS, proinflammatory cytokines, and hypoxia factors [35], which is consistent with patterns from the network analysis (Figure 1).
As mentioned earlier, uremic toxins can be exposed to the circulatory system through the dysbiosis-disrupted leaky mucosal barrier [36]. Mechanistically, uremic toxins and urea-derived metabolites cause degradation of tight junction proteins [37][38], leading to increased translocation of luminal toxic metabolites to the vasculature and kidney. Moreover, mucosal microbiota may be involved in the regulation of xenobiotic metabolic enzymes and transporters in the liver [39][40]. Antibiotic-treated or germ-free animals show altered pharmacokinetics compared with intact hosts, which can be attributed to changes in CYP gene profiles [41]. Microbiota may regulate xenobiotic metabolic enzymes via microbial metabolites that can act as ligands for receptors involved in the induction of genes coding for xenobiotic metabolic enzymes or transporters [39][42]. Microbiota-derived uremic toxins can regulate the expression of genes coding for CYPs and inflammatory mediators via the aryl hydrocarbon receptor (AhR) [42]. Collectively, the PM-induced alterations in the microbiota community contribute to the production and metabolism of URS and PM in the mucosa, which can be translocated to the circulatory system via a disrupted mucosal barrier and has detrimental effects on the renovascular system (Figure 3). Circulating uremic metabolites can also modulate further metabolic and pharmacokinetic processes by regulating the expression and activities of host xenobiotic metabolic enzymes and transporters. Furthermore, PM, URS, and the pool of their metabolites may impact the mucosal barrier, forming a vicious feedback cycle.
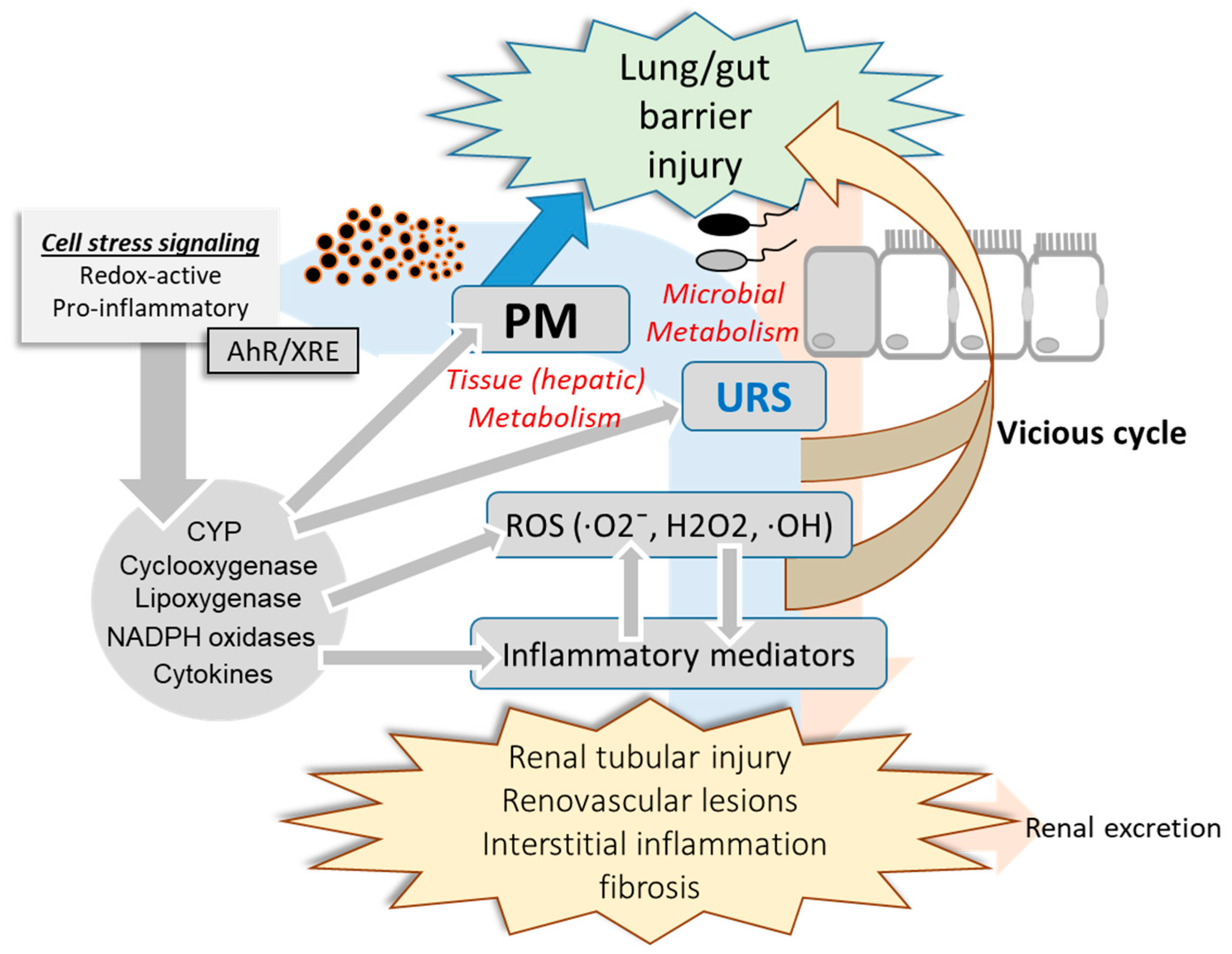
Figure 3. PM-induced alteration in mucosa-kidney axis. PM-insulted mucosal barrier allows the translocation of PM, uremic retention solutes (URS), and their metabolites which can have detrimental effects on the mucosa-kidney axis via stress signaling including AhR-linked pathways. Moreover, invasive microbes and harmful metabolites from the altered microbiota community can contribute to the systemic and renal inflammation during PM exposure. In addition to renal distress, reactive oxygen species (ROS), inflammatory mediators and the circulating xenobiotic agents including PM components and URS injure the mucosal barrier integrity in a feedback way.
5. Mechanistisms of Renal and Vascular Injuries in Response to PM
Circulatory PM and uremic metabolites are detrimental to the renal parenchymal and endothelial tissues in a similar way as the predicted network disruption in the mucosa (Figure 1). In particular, PM can generate ROS including hydroxyl radical (OH) mainly from transition metals and quinones in the airway mucosa [43]. In terms of molecular mechanism of toxicity,∙OH is one of strong genotoxic molecules that quickly bind to and injure DNA [44]. Furthermore, PM-derived redox-active compounds and oxidation products of the lipid membrane can serve as ligands for AhR via transcriptional activation of the xenobiotic responsive element (XRE), leading to expression of genes involved in diverse pathologic events in exposed cells [45]. As previously mentioned, URS and other active microbial metabolites also can contribute to the total pools of AhR ligands during PM exposure. In particular, AhR-XRE signaling mediates expression of ROS-producing metabolic enzymes including cyclooxygenase, lipoxygenase, CYP and NADPH oxidase [46][47][48][49][50][51]. In addition to effects on ROS production and xenobiotic metabolism, AhR-linked signaling is involved in proinflammatory cytokine production and cell death responses [52][53]. URS-induced oxidative stress and proinflammatory cytokines cause necrotic and apoptotic death of the renal tubular and renovascular cells [54][55]. Mechanistically, PM- or URS-activated AhR can disrupt the mitochondrial membrane potential or trigger other diverse cell death signaling pathways, which is the crucial step of renal tubular and renovascular tissue injuries during PM exposure [54][55][56][57]. In contrast, PM, URS, and their active metabolites attenuate the antioxidant capacity in response to the oxidative stress in the mucosa-renal axis. For example, phenyl sulfate, IS and PCS decrease glutathione level in the renal tubular cells [58]. Furthermore, URS-induced chronic distress facilitates progressive interstitial inflammation and renal fibrosis via tissue fibrotic factors including TGF-β1 and α-smooth muscle actin, which ultimately hasten CKD progression [59][60]. Taken together, PM and mucosa-derived metabolites cause renal tubular and endothelial injuries via the oxidative and proinflammatory stress signaling. Moreover, chronic inflammatory and fibrogenic processes aggravate the renal distress during PM exposure.
References
- Mutlu, E.A.; Engen, P.A.; Soberanes, S.; Urich, D.; Forsyth, C.B.; Nigdelioglu, R.; Chiarella, S.E.; Radigan, K.A.; Gonzalez, A.; Jakate, S.; et al. Particulate matter air pollution causes oxidant-mediated increase in gut permeability in mice. Part. Fibre Toxicol. 2011, 8, 19.
- Lee, Y.M.; Lee, J.H.; Kim, H.C.; Ha, E. Effects of PM10 on mortality in pure COPD and asthma-COPD overlap: Difference in exposure duration, gender, and smoking status. Sci. Rep. 2020, 10, 2402.
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2008, 26, 339–362.
- Semmler-Behnke, M.; Takenaka, S.; Fertsch, S.; Wenk, A.; Seitz, J.; Mayer, P.; Oberdorster, G.; Kreyling, W.G. Efficient elimination of inhaled nanoparticles from the alveolar region: Evidence for interstitial uptake and subsequent reentrainment onto airways epithelium. Environ. Health Perspect. 2007, 115, 728–733.
- Li, D.; Li, Y.; Li, G.; Zhang, Y.; Li, J.; Chen, H. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc. Natl. Acad. Sci. USA 2019, 116, 2488–2493.
- Moller, W.; Haussinger, K.; Winkler-Heil, R.; Stahlhofen, W.; Meyer, T.; Hofmann, W.; Heyder, J. Mucociliary and long-term particle clearance in the airways of healthy nonsmoker subjects. J. Appl. Physiol. 2004, 97, 2200–2206.
- Lomer, M.C.; Hutchinson, C.; Volkert, S.; Greenfield, S.M.; Catterall, A.; Thompson, R.P.; Powell, J.J. Dietary sources of inorganic microparticles and their intake in healthy subjects and patients with Crohn’s disease. Br. J. Nutr. 2004, 92, 947–955.
- Lomer, M.C.; Thompson, R.P.; Powell, J.J. Fine and ultrafine particles of the diet: Influence on the mucosal immune response and association with Crohn’s disease. Proc. Nutr. Soc. 2002, 61, 123–130.
- McGill, S.L.; Smyth, H.D. Disruption of the mucus barrier by topically applied exogenous particles. Mol. Pharm. 2010, 7, 2280–2288.
- Derk, R.; Davidson, D.C.; Manke, A.; Stueckle, T.A.; Rojanasakul, Y.; Wang, L. Potential in vitro model for testing the effect of exposure to nanoparticles on the lung alveolar epithelial barrier. Sens. Biosens. Res. 2015, 3, 38–45.
- Zhang, Y.; Hu, H.; Shi, Y.; Yang, X.; Cao, L.; Wu, J.; Asweto, C.O.; Feng, L.; Duan, J.; Sun, Z. (1)H NMR-based metabolomics study on repeat dose toxicity of fine particulate matter in rats after intratracheal instillation. Sci. Total Environ. 2017, 589, 212–221.
- Hong, Z.; Guo, Z.; Zhang, R.; Xu, J.; Dong, W.; Zhuang, G.; Deng, C. Airborne Fine Particulate Matter Induces Oxidative Stress and Inflammation in Human Nasal Epithelial Cells. Tohoku J. Exp. Med. 2016, 239, 117–125.
- Caraballo, J.C.; Yshii, C.; Westphal, W.; Moninger, T.; Comellas, A.P. Ambient particulate matter affects occludin distribution and increases alveolar transepithelial electrical conductance. Respirology 2011, 16, 340–349.
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2012, 9, 35.
- Arrieta, M.C.; Bistritz, L.; Meddings, J.B. Alterations in intestinal permeability. Gut 2006, 55, 1512–1520.
- Kish, L.; Hotte, N.; Kaplan, G.G.; Vincent, R.; Tso, R.; Ganzle, M.; Rioux, K.P.; Thiesen, A.; Barkema, H.W.; Wine, E.; et al. Environmental particulate matter induces murine intestinal inflammatory responses and alters the gut microbiome. PLoS ONE 2013, 8, e62220.
- Dominici, F.; Wang, Y.; Correia, A.W.; Ezzati, M.; Pope, C.A., 3rd; Dockery, D.W. Chemical Composition of Fine Particulate Matter and Life Expectancy: In 95 US Counties Between 2002 and 2007. Epidemiology 2015, 26, 556–564.
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836.
- Rodriguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050.
- Wong, T.Y. Smog induces oxidative stress and microbiota disruption. J. Food Drug Anal. 2017, 25, 235–244.
- Liu, T.; Chen, X.; Xu, Y.; Wu, W.; Tang, W.; Chen, Z.; Ji, G.; Peng, J.; Jiang, Q.; Xiao, J.; et al. Gut microbiota partially mediates the effects of fine particulate matter on type 2 diabetes: Evidence from a population-based epidemiological study. Environ. Int. 2019, 130, 104882.
- Mariani, J.; Favero, C.; Spinazze, A.; Cavallo, D.M.; Carugno, M.; Motta, V.; Bonzini, M.; Cattaneo, A.; Pesatori, A.C.; Bollati, V. Short-term particulate matter exposure influences nasal microbiota in a population of healthy subjects. Environ. Res. 2018, 162, 119–126.
- Li, N.; He, F.; Liao, B.; Zhou, Y.; Li, B.; Ran, P. Exposure to ambient particulate matter alters the microbial composition and induces immune changes in rat lung. Respir. Res. 2017, 18, 143.
- Li, R.; Yang, J.; Saffari, A.; Jacobs, J.; Baek, K.I.; Hough, G.; Larauche, M.H.; Ma, J.; Jen, N.; Moussaoui, N.; et al. Ambient Ultrafine Particle Ingestion Alters Gut Microbiota in Association with Increased Atherogenic Lipid Metabolites. Sci. Rep. 2017, 7, 42906.
- Alderete, T.L.; Jones, R.B.; Chen, Z.; Kim, J.S.; Habre, R.; Lurmann, F.; Gilliland, F.D.; Goran, M.I. Exposure to traffic-related air pollution and the composition of the gut microbiota in overweight and obese adolescents. Environ. Res. 2018, 161, 472–478.
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003.
- Van de Wiele, T.; Vanhaecke, L.; Boeckaert, C.; Peru, K.; Headley, J.; Verstraete, W.; Siciliano, S. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ. Health Perspect. 2005, 113, 6–10.
- Moller, L. In vivo metabolism and genotoxic effects of nitrated polycyclic aromatic hydrocarbons. Environ. Health Perspect. 1994, 102 (Suppl. 4), 139–146.
- Moller, L.; Corrie, M.; Midtvedt, T.; Rafter, J.; Gustafsson, J.A. The role of the intestinal microflora in the formation of mutagenic metabolites from the carcinogenic air pollutant 2-nitrofluorene. Carcinogenesis 1988, 9, 823–830.
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645.
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703.
- Koppe, L.; Fouque, D.; Soulage, C.O. The Role of Gut Microbiota and Diet on Uremic Retention Solutes Production in the Context of Chronic Kidney Disease. Toxins 2018, 10, 155.
- Prokopienko, A.J.; Nolin, T.D. Microbiota-derived uremic retention solutes: Perpetrators of altered nonrenal drug clearance in kidney disease. Expert Rev. Clin. Pharm. 2018, 11, 71–82.
- Lin, C.J.; Liou, T.C.; Pan, C.F.; Wu, P.C.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Wu, C.J. The Role of Liver in Determining Serum Colon-Derived Uremic Solutes. PLoS ONE 2015, 10, e0134590.
- Sun, C.Y.; Hsu, H.H.; Wu, M.S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transpl. 2013, 28, 70–78.
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98.
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043.
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6.
- Liu, T.; Song, X.; Khan, S.; Li, Y.; Guo, Z.; Li, C.; Wang, S.; Dong, W.; Liu, W.; Wang, B.; et al. The gut microbiota at the intersection of bile acids and intestinal carcinogenesis: An old story, yet mesmerizing. Int. J. Cancer 2020, 146, 1780–1790.
- Zhang, J.; Zhang, J.; Wang, R. Gut microbiota modulates drug pharmacokinetics. Drug Metab. Rev. 2018, 50, 357–368.
- Selwyn, F.P.; Cui, J.Y.; Klaassen, C.D. RNA-Seq Quantification of Hepatic Drug Processing Genes in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1572–1580.
- Liu, H.; Narayanan, R.; Hoffmann, M.; Surapaneni, S. The Uremic Toxin Indoxyl-3-Sulfate Induces CYP1A2 In Primary Human Hepatocytes. Drug Metab. Lett. 2016, 10, 195–199.
- Wu, N.; Lu, B.; Chen, J.; Li, X. Size distributions of particle-generated hydroxyl radical (.OH) in surrogate lung fluid (SLF) solution and their potential sources. Environ. Pollut. 2021, 268, 115582.
- Risom, L.; Moller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 2005, 592, 119–137.
- Wang, P.; Thevenot, P.; Saravia, J.; Ahlert, T.; Cormier, S.A. Radical-containing particles activate dendritic cells and enhance Th17 inflammation in a mouse model of asthma. Am. J. Respir. Cell Mol. Biol. 2011, 45, 977–983.
- Harmon, A.C.; Hebert, V.Y.; Cormier, S.A.; Subramanian, B.; Reed, J.R.; Backes, W.L.; Dugas, T.R. Particulate matter containing environmentally persistent free radicals induces AhR-dependent cytokine and reactive oxygen species production in human bronchial epithelial cells. PLoS ONE 2018, 13, e0205412.
- Pardo, M.; Shafer, M.M.; Rudich, A.; Schauer, J.J.; Rudich, Y. Single Exposure to near Roadway Particulate Matter Leads to Confined Inflammatory and Defense Responses: Possible Role of Metals. Environ. Sci. Technol. 2015, 49, 8777–8785.
- Kopf, P.G.; Walker, M.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases reactive oxygen species production in human endothelial cells via induction of cytochrome P4501A1. Toxicol. Appl. Pharm. 2010, 245, 91–99.
- Costa, C.; Catania, S.; De Pasquale, R.; Stancanelli, R.; Scribano, G.M.; Melchini, A. Exposure of human skin to benzopyrene: Role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicology 2010, 271, 83–86.
- Degner, S.C.; Papoutsis, A.J.; Selmin, O.; Romagnolo, D.F. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3’-diindolylmethane in breast cancer cells. J. Nutr. 2009, 139, 26–32.
- Su, H.H.; Lin, H.T.; Suen, J.L.; Sheu, C.C.; Yokoyama, K.K.; Huang, S.K.; Cheng, C.M. Aryl hydrocarbon receptor-ligand axis mediates pulmonary fibroblast migration and differentiation through increased arachidonic acid metabolism. Toxicology 2016, 370, 116–126.
- Umannova, L.; Zatloukalova, J.; Machala, M.; Krcmar, P.; Majkova, Z.; Hennig, B.; Kozubik, A.; Vondracek, J. Tumor necrosis factor-alpha modulates effects of aryl hydrocarbon receptor ligands on cell proliferation and expression of cytochrome P450 enzymes in rat liver “stem-like” cells. Toxicol. Sci. 2007, 99, 79–89.
- Huang, Y.; He, J.; Liang, H.; Hu, K.; Jiang, S.; Yang, L.; Mei, S.; Zhu, X.; Yu, J.; Kijlstra, A.; et al. Aryl Hydrocarbon Receptor Regulates Apoptosis and Inflammation in a Murine Model of Experimental Autoimmune Uveitis. Front. Immunol. 2018, 9, 1713.
- Cheng, T.H.; Ma, M.C.; Liao, M.T.; Zheng, C.M.; Lu, K.C.; Liao, C.H.; Hou, Y.C.; Liu, W.C.; Lu, C.L. Indoxyl Sulfate, a Tubular Toxin, Contributes to the Development of Chronic Kidney Disease. Toxins 2020, 12, 684.
- Lano, G.; Burtey, S.; Sallee, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229.
- Boovarahan, S.R.; Kurian, G.A. Mitochondrial dysfunction: A key player in the pathogenesis of cardiovascular diseases linked to air pollution. Rev. Environ. Health 2018, 33, 111–122.
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Lo Guidice, J.M.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449.
- Edamatsu, T.; Fujieda, A.; Itoh, Y. Phenyl sulfate, indoxyl sulfate and p-cresyl sulfate decrease glutathione level to render cells vulnerable to oxidative stress in renal tubular cells. PLoS ONE 2018, 13, e0193342.
- Saito, S.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Enomoto, A.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor is involved in expression of TGF-beta1 and alpha-smooth muscle actin in proximal tubular cells. Endocrinology 2014, 155, 1899–1907.
- Milanesi, S.; Garibaldi, S.; Saio, M.; Ghigliotti, G.; Picciotto, D.; Ameri, P.; Garibotto, G.; Barisione, C.; Verzola, D. Indoxyl Sulfate Induces Renal Fibroblast Activation through a Targetable Heat Shock Protein 90-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2050183.
More
Information
Subjects:
Urology & Nephrology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
488
Revisions:
2 times
(View History)
Update Date:
31 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No