Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrii P. Gryganskyi | -- | 3025 | 2023-07-26 16:10:22 | | | |
| 2 | Dean Liu | -16 word(s) | 3009 | 2023-07-28 05:24:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gryganskyi, A.P.; Golan, J.; Muszewska, A.; Idnurm, A.; Dolatabadi, S.; Mondo, S.J.; Kutovenko, V.B.; Kutovenko, V.O.; Gajdeczka, M.T.; Anishchenko, I.M.; et al. Genomes and the Evolution of the Zygomycetes. Encyclopedia. Available online: https://encyclopedia.pub/entry/47318 (accessed on 26 July 2026).
Gryganskyi AP, Golan J, Muszewska A, Idnurm A, Dolatabadi S, Mondo SJ, et al. Genomes and the Evolution of the Zygomycetes. Encyclopedia. Available at: https://encyclopedia.pub/entry/47318. Accessed July 26, 2026.
Gryganskyi, Andrii P., Jacob Golan, Anna Muszewska, Alexander Idnurm, Somayeh Dolatabadi, Stephen J. Mondo, Vira B. Kutovenko, Volodymyr O. Kutovenko, Michael T. Gajdeczka, Iryna M. Anishchenko, et al. "Genomes and the Evolution of the Zygomycetes" Encyclopedia, https://encyclopedia.pub/entry/47318 (accessed July 26, 2026).
Gryganskyi, A.P., Golan, J., Muszewska, A., Idnurm, A., Dolatabadi, S., Mondo, S.J., Kutovenko, V.B., Kutovenko, V.O., Gajdeczka, M.T., Anishchenko, I.M., Pawlowska, J., Tran, N.V., Ebersberger, I., Voigt, K., Wang, Y., Chang, Y., Pawlowska, T.E., Heitman, J., Vilgalys, R., ...Stajich, J.E.. (2023, July 26). Genomes and the Evolution of the Zygomycetes. In Encyclopedia. https://encyclopedia.pub/entry/47318
Gryganskyi, Andrii P., et al. "Genomes and the Evolution of the Zygomycetes." Encyclopedia. Web. 26 July, 2023.
Copy Citation
The first genome sequence of a zygomycete fungus was from a clinical strain of Rhizopus delemar that was isolated from a patient with mucormycosis—a highly destructive and lethal infection that is typically seen in immunocompromised hosts. Sequencing of the R. delemar genome was driven from the clinical perspective, but it also provided the first genome of a fungus outside of the Dikarya.
fungal evolution
ecological relevance
pathogens
model organisms
1. Mucoromycota
Mucoromycota comprises many ubiquitous soil saprotrophs, among them critically important plant symbionts, including the arbuscular mycorrhizal fungi (Glomeromycotina) [1], Endogonales (Mucoromycotina) [2], “fine root endophytes” (Mucoromycotina) [3], and putative root endophytic species including Mortierella (Mortierellomycotina) [4], Pygmaeomycetaceae (Mucoromycotina) [5], and Umbelopsis (Mucoromycotina) [6]. On the other hand, many fungi within this group are also noted plant pathogens (e.g., Rhizopus microsporus, which causes rice seedling blight, and various Choanephora, Gilbertella, and Mucor species that cause post-harvest rots) as well as numerous species (Actinomucor, Apophysomyces, Cokeromyces, Cunninghamella, Lichtheimia, Mucor, Rhizomucor, Rhizopus, Saksenea, Syncephalastrum) that can cause the disease mucormycosis in humans and other animals [7].
Most zygomycete genomes are of species within Mucoromycota; as a result, much of what resesarchers know about genome evolution among early-diverging fungi comes from this phylum. Genome duplication appears to have played a significant role in their evolution, as first described for Rhizopus delemar and Mucor circinelloides [8][9]. Genome sequencing has also revealed a diversity of endobacteria that may be found as symbionts in the Mucoromycota, e.g., in Rhizopus microsporus and Endogone pisiformis (Mucoromycotina), Linnemannia elongata and Mortierella alpina (Mortierellomycotina), and Rhizophagus irregularis (Glomeromycotina) [10][11][12][13][14][15]. Many plant-associated fungal lineages contain endobacteria, but the potential role of these symbionts in the land colonization process remain to be characterized. “Foreign” DNA in the form of diverse mycoviruses has also been frequently detected during genome sequencing of zygomycetes [16][17]. Genomics enabled the systematic study of genome architecture [18][19][20], genetic manipulations [21], and intra-isolate genomic variations [22]. Genome-wide searches and comparisons have also detected evidence for transposon spread through genomes [23][24][25] and uniquely high 6-methyladenine DNA modifications in zygomycete fungi as compared to Dikarya [26]. Studies on metabolic patterns across the fungal kingdom, the search for the mating gene loci and protein families [27][28][29], and the phylogenies of certain taxonomic groups [30][31] have also been conducted using-whole genome sequencing data. Genome-based research has also provided insights into the trophic modes of zygomycete fungi, including the evolution of mycorrhizae [2][32] and commonalities in the pathogenicity of Mucoralean fungi [33][34].
2. Mucoromycotina
Genome sequencing of this subphylum has been the most comprehensive among all the basal fungal lineages although genomes are still unavailable for several genera, including species of Siepmannia, Chlamydoabsidia, and Utharomyces, as well as for two new root-associated species of Pygmaeomyces of the new genus and family Pygmaeomycetaceae [5]. Furthermore, the plant-associated order Endogonales remains highly undersampled despite recent advances [2][35]. Some of its “fine root endophytes” colonize early-diverging plants, and sometimes co-occur with AMF [36][37]. The average genome size in Mucoromycotina species ranges from 35 to 50 Mbp, with species of Umbelopsis at the low end of the spectrum (22–30 Mbp) and Endogonales such as Jimgerdemannia species at the higher end (240 Mbp). For some genera, several hundred genomes have already been sequenced. For example, there are >220 genomes available for species of Rhizopus, mostly of R. arrhizus (syn. R. oryzae) and its varieties and synonyms, and of R. microsporus, R. delemar, and R. stolonifer. Rhizopus is increasingly studied as a model organism thanks to its availability in culture collections and its important role in biotechnology and human health. Genomic information might be helpful for future studies of pathogenicity in Rhizopus species since infections by members of this genus are the leading cause of human mucormycosis [30][33]. Phylogenomic data suggest that species of Rhizopus are part of a well-resolved monophyletic group. However, this is in stark contrast to the situation among members of the genus Mucor (>150 genomes), which is incredibly diverse and is likely polyphyletic (Figure 1). The additional Mucoromycotina genomes consist mostly of saprotrophic species that are relevant to human health (species of Lichtheimia and Cunninghamella) and biotechnology (Phycomyces blakesleeanus) [38][39][40]. Genomes of Endogonales (as well as Glomeromycotina) contain effectors to facilitate plant–fungus interactions, which apparently aided in the terrestrialization of Earth by early land plants. Genome information helped to describe and clarify the phylogenetic placement of new lineages and species, such as Bifiguratus [41] and Calcarisporiella [42].
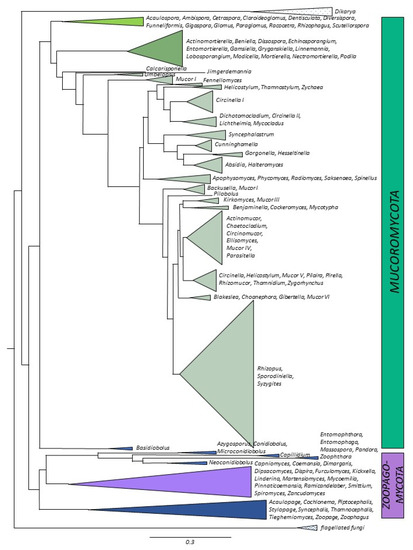
Figure 1. Distributions of the number of genomes sequenced imposed onto phylogenetic tree. Different shades of green and brown—Mucoromycota, purple and blue—Zoopagomycota.
3. Mortierellomycotina
This subphylum contains an estimated 170 species [43] and was recently circumscribed into 13 monophyletic genera using multi-locus and low-coverage genome sequencing [44]. Reference genome sequences of both low quality and high quality are now available for representatives for most genera, including Actinomortierella, Benniella, Dissophora, Entomortierella, Gamsiella, Gryganskiella, Linnemannia, Lobosporangium, Lunasporangiospora, Mortierella, Necromortierella, Podila, and the sporocarpic genus Modicella [44][45]. A high diversity of Mortierellomycotina species can be isolated from soil, but it appears that genera may differ in their ecological roles. For example, Necromortierella species appear to be necrotrophic, whereas Actinomortierella spp. are often associated with millipedes, and Entomortierella spp. are often associated with arthropods [46]. Many genera, such as Linnemannia and Podila, have been recovered as rhizosphere associates and endophytes of plant roots [47][48], and they appear to improve plant growth and impact flower and seed production [49]. Several species such as Mortierella alpina have been sequenced due to their use in industry and biotechnology for lipid production [50].
4. Glomeromycotina
Arbuscular mycorrhizal fungi (AMFs) are arguably the most common and oldest symbionts of terrestrial plants [1], credited with their role in the facilitation of plant transition to the terrestrial habitat [51]. AMFs supply their plant hosts with mineral nutrients translocated from the soil in return for plant-assimilated carbon [52]. Yet, there are not many genomic sequences of these organisms available due to historical impediments hampering sequencing projects, including multinucleate spores with different genotypes, obligate biotrophy of AMFs, and the initial uncertainty concerning their genome sizes, with conflicting size estimates in Rhizophagus irregularis ranging from 14.1 [53] to 154.8 Mb [54]. Once these obstacles were surmounted, genomic data confirmed that AMFs have unusually large genomes, ranging from 153 Mb in R. irregularis [55] to 773.1 Mb in Gigaspora margarita [56]. This size enhancement relative to other Mucoromycota is driven by lineage-specific expansions of gene families and massive proliferation of transposable elements [32][55][56]. Genomic data also revealed that AMFs display several other features that differentiate them not only from Mucoromycota but also from other fungi. For example, despite the lack of morphological evidence of sexual reproduction, the R. irregularis genome harbors a candidate mating-type locus with two genes encoding homeodomain-like transcription factors [57]. If these homeodomain-like genes indeed control sex, then regulation of the reproductive processes in AMFs would resemble mating in Dikarya rather than in Mucoromycota, which rely on HMG domain proteins for sex determination [58]. Rhizophagus irregularis differs also from other fungi in the organization of its rRNA operons. Instead of being tandemly arrayed as they are in other fungi, the rRNA operons in Glomeromycotina are dispersed individually across the genome [55]. This important discovery put an end to a long-lasting debate on whether intraindividual rRNA gene variation typical for AMFs is distributed among dissimilar nuclei [59] or contained in each nucleus [60], supporting the latter scenario. Lastly, genome-based inferences of metabolic capacity revealed that AMFs lack the gene encoding fatty acid synthase [61][62][63], which makes them obligately dependent on plant hosts for lipids [64][65][66][67]. As in other fastidious organisms, this information is important for devising cultivation strategies that complement fatty acid auxotrophy of AMF [68][69]. Even though accumulation of genomic data for AMF has lagged behind other Mucoromycota, the insights gathered so far have been critical for resolving several puzzles in the biology of these unique organisms. PacBio HiFi and Hi-C sequencing of all available AMF heterokaryons helped to discover the two sets of coexisting homologous chromosomes in one heterokaryon. Genes required for plant colonization were found in gene-sparse, repeat-reach compartments [70]. Another important question that remains to be addressed is which genomic features of AMFs contribute to superior yield outcomes in agronomic crop species, as AMFs do not universally benefit all plant hosts.
5. Zoopagomycota
In contrast to Mucoromycota, most species of Zoopagomycota are parasites or pathogens of animals and fungi. This includes the insect pathogenic group Entomophthoromycotina (some members of which exhibit behavioral control over their hosts) [71], the Kickxellomycotina which includes a mixture of ecologies (including commensalistic arthropods gut fungi) [72], and Zoopagomycotina, which are parasites of fungi (mainly in Mucoromycota) and microinvertebrates (e.g., amoebae, nematodes, rotifers) [73]. There are also opportunistic human pathogens in this phylum, including species of Basidiobolus and Conidiobolus [74]. Axenic culturing of some fungi in Zoopagomycota is possible [75][76] but extracting high-quality genomic DNA from many species remains difficult. Sometimes there is a need to extract the genomic material of the parasite together with the host (often another fungus or insect) and separate their genomes during assembly, which creates additional challenges. Small amounts of available DNA have necessitated the use of single-cell DNA extraction in some cases, especially in species with dramatically reduced vegetative mycelial structures. Much greater success has been achieved for putative saprotrophic fungi such as dung-inhabiting species of Kickxellales. Genomes and transcriptomes have been used primarily to resolve various evolutionary questions, including the monophyletic origin of Zoopagales [73], the evolution of ploidy from a diploid ancestral stage [77], and loss of a large number of pectinases and other plant cell-wall-degrading enzymes in Zoopagomycota [78]. Genome data were also successfully used to resolve evolutionary relationships within particular taxonomic groups, such as Massospora [46], to track other evolutionary events such as horizontal gene transfer events in gut fungi [79], and to reveal the genetic toolbox in insect symbionts [80]. These studies help researchers to understand the evolutionary trajectories of genome size, namely, a dramatic increase in genome size for the insect pathogens and insect endosymbionts as compared to the genome size in the saprobes and mycoparasites. Genome data have also helped to clarify fundamental differences among Zoopagomycota lineages in their production of secondary metabolites [81], as well as metabolic pathways in general [77]. Mating genetics in this phylum is still not well understood, and that is a question that remains to be addressed with genomic data.
6. Kickxellomycotina
This subphylum contains an interesting mix of mycoparasites (Dimargaritales), putative saprotrophs (Kickxellales), and obligate insect endobionts (Asselariales, Barbatosporales, Harpellales, and Orphellales). The Dimargaritales are similar to mycoparasites in the Zoopagales in that they form penetration hyphae (haustoria) and mostly attack similar host species in the Mucoromycota. In contrast to the Zoopagales mycoparasites, species of Dimargaritales appear to be rare in the environment and have mostly been isolated from dung rather than soil. Recent genomic analyses have also highlighted that members of the Dimargaritales that have larger genomes and more predicted secondary metabolite genes compared to their close relatives [72][81]. Furthermore, single-cell genome data have suggested that Dimargaris cristalligena may be nonhaploid and missing certain enzymes from metabolic pathways (e.g., thiamine biosynthesis and biotin metabolism) [77]. Identifying such genomic features of symbiotic species can provide targets for further experimental tests to understand the molecular interactions between hosts and parasites.
On the other hand, the Kickxellales is the only group of Zoopagomycota containing mostly saprotrophic species, and many appear to be rare in the environment, having only been reported in the literature one or a few times. An exception is the genus Coemansia, the species of which are commonly isolated from soil and dung samples [82][83]. Genome sequencing efforts have demonstrated that Kickxellales species have small genomes (less than 40 MB) and variability in their predicted secondary metabolite genes, with Coemansia and Linderina, along with more polyketide synthases than other Kickxellales and D. cristalligena having more nonribosomal peptide synthetases than other Kickxellales [81]. A preliminary rDNA phylogeny showed polyphyly among Kickxellales [83], and a large-scale sequencing project that utilized low-coverage genome data from >100 isolates to test these hypotheses, as well as explore the diversity of Coemansia species, resolved many of these relationships [72].
The gut fungi (Trichomycetes) include arthropod symbionts for which several draft genomes have been available since 2016 [84]. These fungi are distinct from the saprotrophic and coprophilic relatives (i.e., Kickxellales) and belong to four distinct phylogenetic lineages—Asellariales, Barbatosporales, Harpellales, and Orphellales [85]. Asellariales are associated with isopods and springtails (Collembola) [86]. Species within the three genera (Asellaria, Baltomyces, and Orchesellaria) have been recovered from both terrestrial and aquatic hosts [86], but their phylogenetic relationships remain unclear, as sequence data have only been obtained from Asellaria [85], and no cultures are available for this group.
Harpellales species are usually found in lower Diptera insect larvae and have coevolved with their hosts for over 200 million years [87]. Species of Orphellales are stonefly nymph symbionts with unusual morphological characters (i.e., asexual and sexual spore shapes, and thalli protruding beyond the anus of insect hosts) [88], which cluster together with other Trichomycetes and the Spiromycetales for a sister clade to Kickxellales [72]. Smittium is a well-studied genus within Harpellales with approximately 100 described species [89]. However, there are only nine whole-genome sequences available for these insect gut-dwelling fungi, and all were made from culturable Harpellales species, with the best assembly—Capniomyces stellatus—having 72 scaffolds [84]. Barbatosporales are monotypic, and the single species (Barbatospora ambicaudata) has only been recorded from black fly larvae collected in the Great Smoky Mountains National Park in Tennessee, obtained in axenic culture [90].
Although researchers only have a handful of Harpellales genomes, they have already expanded the knowledge and understanding of these enigmatic microbial fungi in multiple ways. For example, a mosquito-like polyubiquitin gene has been identified in Zancudomyces culisetae (Harpellales), which was presumably acquired via a horizontal gene transfer event [79]. Comparative genomics revealed a fungus–insect symbiotic core gene toolbox (FISCoG) that is shared among the insect-associated fungi (e.g., Harpellales, Basidiobolus, and Conidiobolus) and higher-ranked Hypocreales (Ascomycota). However, the insect pathogens (Hypocreales and Entomophthoromycotina members) tend to have genomes enriched in genes that are useful for pathogenic processes such as the platelet-activating factor acetyl-hydrolase coding genes, whereas the gut commensals have genomes enriched in cell adhesion genes for a successful gut-dwelling lifestyle [80]. In addition, Harpellales genomes also facilitated a kingdom-wide study to confirm the production of selenoproteins in early-diverging fungal lineages [91]. As a major group of early-diverging fungi, representing seven of the nine fungal species that utilize selenoproteins, Harpellales may take the advantages of selenocysteine over cysteine for specific oxidoreductase functions.
Unfortunately, no efforts have been successful in obtaining genome information of the Asellariales, Barbatosporales, and Orphellales, which colonize different host types. However, multigene analyses incorporating low-coverage genome data (along with data from Asellariales and Orphellales) suggested an alternate hypothesis for the evolution of fungal–insect associations. Contrary to previous topologies that suggested multiple events [85], the larger dataset showed a possible single origin of gut fungi [72]. Further evolutionary evidence may be identified with the help of their genome sequences to understand how these gut-dwelling microbial fungi interact with various aquatic insect hosts and evaluate the origins of the symbiosis.
7. Zoopagomycotina
Zoopagomycotina species are fascinating, partially due to their cryptic ecological niches and ability to parasitize various hosts such as amoebae, nematodes, rotifers, and other fungi. It is still challenging to culture most of the Zoopagomycotina isolates. Thus, it is not surprising to see that Zoopagomycotina has the lowest number of available genomes, the qualities of which are not comparable to other lineages, especially the well-sequenced Mucoromycotina. Recently developed single-cell genomic techniques have provided an alternative, culture-independent method to obtain their genomic information [77]. On the basis of single-cell or multiple-cell libraries, the completeness of genome assemblies can be as high as 89.6% (Piptocephalis tieghemiana RSA 1565) [73]. The assembled Zoopagomycotina genomes indicate that this clade of fungi is characterized by small genome size, but the numbers of protein-coding genes are not necessarily low. Some of the genomes can encode close to 10,000 genes (e.g., Zoophagus insidians, a predator of rotifers and nematodes. It seems likely that these completely microscopic fungi are capable of more complicated and yet-to-be-identified interactions with their animal and/or fungal hosts, but more work will be needed to explore these interactions.
Two additional Zoopagales have remarkably small genomes: a species of Acaulopage (11 Mbp) and a species of Zoopage (14 Mbp). No genomes have yet been published for the following genera: Amoebophilus, Aplectosoma, Bdellospora, Brachymyces, Cystopage, Endocochlus, Euryancale, Helicocephalum, Reticulocephalis, and Sigmoideomyces.
8. Entomophthoromycotina
This lineage remains the most insufficiently sequenced fungal group at the subphylum level. Despite many genomes from the genus Conidiobolus, there are only a few other species (all from the key family Entomophthoraceae) for which genomes are available. The genus Conidiobolus is a polyphyletic assemblage of multiple lineages. This group of Conidiobolus-like fungi was recently phylogenetically divided into three new families, using a combined multi-gene and phylogenomic approach, revealing that the ballistic conidia arose prior to their transition to a parasitic lifestyle [31][92]. Difficulties with culturing and, therefore, obtaining enough high-molecular-weight genomic DNA represent a reason why some species only have transcriptomes available. Alternatively, in some cases, the fungal pathogen is sequenced together with its host, and then the reads are separated bioinformatically. Aside from the polyphyletic genus Conidiobolus and entomophthoroid-like Basidiobolus, genome or transcriptome data are available only for a few species of Entomophaga, Entomophthora, Massospora, Pandora, and Zoophthora. No genomes yet exist for Ancylistes, Apterivorax, Batkoa, Erynia, Eryniopsis, Furia, Macrobiotophthora, Meristacrum, Neozygites, Orthomyces, Tabanomyces, Thaxterosporium, Schizangiella, and Strongwellsea. The largest genomes recovered are over 2000 Mbp in Massospora and 650 Mbp in Zoophthora, with possibly similar genome sizes in Entomophthora and Entomophaga. A phylogenetic analysis recently demonstrated that Basidiobolus, a genus of amphibian gut-dwelling fungi, does not belong to Entomophthorales, where it was traditionally placed, mostly on the basis of its production of forcibly discharged conidia that are morphologically similar to those in other entomophthoralean fungi [93]. The genus was difficult to place with rDNA data [94], but was inferred either as a separate clade sister to the Mucoromycota [95] or as the earliest diverging lineage within Zoopagomycota [80], on the basis of phylogenomic data. The ballistic spore dispersal mechanism, which differs structurally from that found in the Entomophthorales, is likely an example of convergent evolution among fungi that appear to be similar but are distantly related. In addition to being evolutionarily enigmatic, the genomes of B. meristosporus and B. heterosporus are larger than most other basal fungi and contained a much higher proportion of secondary metabolite-encoding genes than other Zoopagomycota species that have been studied thus far. Furthermore, Basidiobolus genomes contain numerous genes that putatively originated from horizontal gene transfer events from bacteria [81].
References
- Genre, A.; Lanfranco, L.; Perotto, S.; Bonfante, P. Unique and Common Traits in Mycorrhizal Symbioses. Nat. Rev. Microbiol. 2020, 18, 649–660.
- Chang, Y.; Desirò, A.; Na, H.; Sandor, L.; Lipzen, A.; Clum, A.; Barry, K.; Grigoriev, I.V.; Martin, F.M.; Stajich, J.E.; et al. Phylogenomics of Endogonaceae and Evolution of Mycorrhizas within Mucoromycota. New Phytol. 2019, 222, 511–525.
- Hoysted, G.A.; Jacob, A.S.; Kowal, J.; Giesemann, P.; Bidartondo, M.I.; Duckett, J.G.; Gebauer, G.; Rimington, W.R.; Schornack, S.; Pressel, S.; et al. Mucoromycotina Fine Root Endophyte Fungi Form Nutritional Mutualisms with Vascular Plants. Plant Physiol. 2019, 181, 565–577.
- Wani, Z.A.; Kumar, A.; Sultan, P.; Bindu, K.; Riyaz-Ul-Hassan, S.; Ashraf, N. Mortierella alpina CS10E4, an Oleaginous Fungal Endophyte of Crocus sativus L. Enhances Apocarotenoid Biosynthesis and Stress Tolerance in the Host Plant. Sci. Rep. 2017, 7, 8598.
- Walsh, E.; Luo, J.; Khiste, S.; Scalera, A.; Sajjad, S.; Zhang, N. Pygmaeomycetaceae, a New Root-Associated Family in Mucoromycotina from the Pygmy Pine Plains. Mycologia 2021, 113, 134–145.
- Liu, S.; Liu, M.; Liao, Q.-G.; Lü, F.-B.; Zhao, X.-L. Effects of Inoculated Mycorrhizal Fungi and Non-Mycorrhizal Beneficial Micro-Organisms on Plant Traits, Nutrient Uptake and Root-Associated Fungal Community Composition of the Cymbidium hybridum in Greenhouse. J. Appl. Microbiol. 2021, 131, 413–424.
- Steinbrink, J.M.; Miceli, M.H. Clinical Review of Mucormycosis. Infect. Dis. Clin. N. Am. 2021, 35, 435–452.
- Ma, L.-J.; Ibrahim, A.S.; Skory, C.; Grabherr, M.G.; Burger, G.; Butler, M.; Elias, M.; Idnurm, A.; Lang, B.F.; Sone, T.; et al. Genomic Analysis of the Basal Lineage Fungus Rhizopus oryzae Reveals a Whole-Genome Duplication. PLoS Genet. 2009, 5, e1000549.
- Corrochano, L.M.; Kuo, A.; Marcet-Houben, M.; Polaino, S.; Salamov, A.; Villalobos-Escobedo, J.M.; Grimwood, J.; Álvarez, M.I.; Avalos, J.; Bauer, D.; et al. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016, 26, 1577–1584.
- Partida-Martinez, L.P.; Hertweck, C. Pathogenic Fungus Harbours Endosymbiotic Bacteria for Toxin Production. Nature 2005, 437, 884–888.
- Torres-Cortés, G.; Ghignone, S.; Bonfante, P.; Schüßler, A. Mosaic Genome of Endobacteria in Arbuscular Mycorrhizal Fungi: Transkingdom Gene Transfer in an Ancient Mycoplasma-Fungus Association. Proc. Natl. Acad. Sci. USA 2015, 112, 7785–7790.
- Mondo, S.J.; Salvioli, A.; Bonfante, P.; Morton, J.B.; Pawlowska, T.E. Nondegenerative Evolution in Ancient Heritable Bacterial Endosymbionts of Fungi. Mol. Biol. Evol. 2016, 33, 2216–2231.
- Bonfante, P.; Desirò, A. Who Lives in a Fungus? The Diversity, Origins and Functions of Fungal Endobacteria Living in Mucoromycota. ISME J. 2017, 11, 1727–1735.
- Uehling, J.; Gryganskyi, A.; Hameed, K.; Tschaplinski, T.; Misztal, P.K.; Wu, S.; Desirò, A.; Vande Pol, N.; Du, Z.; Zienkiewicz, A.; et al. Comparative Genomics of Mortierella elongata and Its Bacterial Endosymbiont Mycoavidus cysteinexigens. Environ. Microbiol. 2017, 19, 2964–2983.
- Lastovetsky, O.A.; Ahn, E.; Mondo, S.J.; Toomer, K.H.; Zhang, A.; Johnson, L.M.; Pawlowska, T.E. Distribution and Population Structure of Endobacteria in Arbuscular Mycorrhizal Fungi at North Atlantic Dunes. ISME J. 2018, 12, 3001–3013.
- Myers, J.M.; Bonds, A.E.; Clemons, R.A.; Thapa, N.A.; Simmons, D.R.; Carter-House, D.; Ortanez, J.; Liu, P.; Miralles-Durán, A.; Desirò, A.; et al. Survey of Early-Diverging Lineages of Fungi Reveals Abundant and Diverse Mycoviruses. mBio 2020, 11, e02027-20.
- Espino-Vázquez, A.N.; Bermúdez-Barrientos, J.R.; Cabrera-Rangel, J.F.; Córdova-López, G.; Cardoso-Martínez, F.; Martínez-Vázquez, A.; Camarena-Pozos, D.A.; Mondo, S.J.; Pawlowska, T.E.; Abreu-Goodger, C.; et al. Narnaviruses: Novel Players in Fungal-Bacterial Symbioses. ISME J. 2020, 14, 1743–1754.
- Bever, J.D.; Kang, H.-J.; Kaonongbua, W.; Wang, M. Genomic Organization and Mechanisms of Inheritance in Arbuscular Mycorrhizal Fungi: Contrasting the Evidence and Implications of Current Theories. In Mycorrhiza; Varma, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 135–148. ISBN 978-3-540-78824-9.
- Martin, F.; Gianinazzi-Pearson, V.; Hijri, M.; Lammers, P.; Requena, N.; Sanders, I.R.; Shachar-Hill, Y.; Shapiro, H.; Tuskan, G.A.; Young, J.P.W. The Long Hard Road to a Completed Glomus intraradices Genome. New Phytol. 2008, 180, 747–750.
- Lebreton, A.; Corre, E.; Jany, J.-L.; Brillet-Guéguen, L.; Pèrez-Arques, C.; Garre, V.; Monsoor, M.; Debuchy, R.; Le Meur, C.; Coton, E.; et al. Comparative Genomics Applied to Mucor Species with Different Lifestyles. BMC Genom. 2020, 21, 135.
- Vellanki, S.; Navarro-Mendoza, M.I.; Garcia, A.E.; Murcia, L.; Perez-Arques, C.; Garre, V.; Nicolas, F.E.; Lee, S.C. Mucor circinelloides: Growth, Maintenance and Genetic Manipulation. Curr. Protoc. Microbiol. 2018, 49, e53.
- Boon, E.; Zimmerman, E.; Lang, B.F.; Hijri, M. Intra-Isolate Genome Variation in Arbuscular Mycorrhizal Fungi Persists in the Transcriptome. J. Evol. Biol. 2010, 23, 1519–1527.
- Muszewska, A.; Okrasińska, A.; Steczkiewicz, K.; Drgas, O.; Orłowska, M.; Perlińska-Lenart, U.; Aleksandrzak-Piekarczyk, T.; Szatraj, K.; Zielenkiewicz, U.; Piłsyk, S.; et al. Metabolic Potential, Ecology and Presence of Associated Bacteria Is Reflected in Genomic Diversity of Mucoromycotina. Front. Microbiol. 2021, 12, 636986.
- Muszewska, A.; Steczkiewicz, K.; Ginalski, K. DIRS and Ngaro Retrotransposons in Fungi. PLoS ONE 2013, 8, e76319.
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Cut-and-Paste Transposons in Fungi with Diverse Lifestyles. Genome Biol. Evol. 2017, 9, 3463–3477.
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread Adenine N6-Methylation of Active Genes in Fungi. Nat. Genet. 2017, 49, 964–968.
- Corradi, N.; Sanders, I.R. Evolution of the P-Type II ATPase Gene Family in the Fungi and Presence of Structural Genomic Changes among Isolates of Glomus intraradices. BMC Evol. Biol. 2006, 6, 21.
- Halary, S.; Malik, S.-B.; Lildhar, L.; Slamovits, C.H.; Hijri, M.; Corradi, N. Conserved Meiotic Machinery in Glomus spp., a Putatively Ancient Asexual Fungal Lineage. Genome Biol. Evol. 2011, 3, 950–958.
- Halary, S.; Daubois, L.; Terrat, Y.; Ellenberger, S.; Wöstemeyer, J.; Hijri, M. Mating Type Gene Homologues and Putative Sex Pheromone-Sensing Pathway in Arbuscular Mycorrhizal Fungi, a Presumably Asexual Plant Root Symbiont. PLoS ONE 2013, 8, e80729.
- Gryganskyi, A.P.; Golan, J.; Dolatabadi, S.; Mondo, S.; Robb, S.; Idnurm, A.; Muszewska, A.; Steczkiewicz, K.; Masonjones, S.; Liao, H.-L.; et al. Phylogenetic and Phylogenomic Definition of Rhizopus Species. G3 (Bethesda) 2018, 8, 2007–2018.
- Gryganskyi, A.P.; Nie, Y.; Hajek, A.E.; Hodge, K.T.; Liu, X.-Y.; Aadland, K.; Voigt, K.; Anishchenko, I.M.; Kutovenko, V.B.; Kava, L.; et al. The Early Terrestrial Fungal Lineage of Conidiobolus-Transition from Saprotroph to Parasitic Lifestyle. J. Fungi 2022, 8, 789.
- Tisserant, E.; Malbreil, M.; Kuo, A.; Kohler, A.; Symeonidi, A.; Balestrini, R.; Charron, P.; Duensing, N.; Frei dit Frey, N.; Gianinazzi-Pearson, V.; et al. Genome of an Arbuscular Mycorrhizal Fungus Provides Insight into the Oldest Plant Symbiosis. Proc. Natl. Acad. Sci. USA 2013, 110, 20117–20122.
- Chibucos, M.C.; Soliman, S.; Gebremariam, T.; Lee, H.; Daugherty, S.; Orvis, J.; Shetty, A.C.; Crabtree, J.; Hazen, T.H.; Etienne, K.A.; et al. An Integrated Genomic and Transcriptomic Survey of Mucormycosis-Causing Fungi. Nat. Commun. 2016, 7, 12218.
- Wang, Y.; Chang, Y.; Ortañez, J.; Peña, J.F.; Carter-House, D.; Reynolds, N.K.; Smith, M.E.; Benny, G.; Mondo, S.J.; Salamov, A.; et al. Divergent Evolution of Early Terrestrial Fungi Reveals the Evolution of Mucormycosis Pathogenicity Factors. Genome Biol. Evol. 2023, 15, evad046.
- Desirò, A.; Rimington, W.R.; Jacob, A.; Pol, N.V.; Smith, M.E.; Trappe, J.M.; Bidartondo, M.I.; Bonito, G. Multigene Phylogeny of Endogonales, an Early Diverging Lineage of Fungi Associated with Plants. IMA Fungus 2017, 8, 245–257.
- Ogura-Tsujita, Y.; Yamamoto, K.; Hirayama, Y.; Ebihara, A.; Morita, N.; Imaichi, R. Fern Gametophytes of Angiopteris lygodiifolia and Osmunda japonica Harbor Diverse Mucoromycotina Fungi. J. Plant Res. 2019, 132, 581–588.
- Rimington, W.R.; Pressel, S.; Duckett, J.G.; Field, K.J.; Bidartondo, M.I. Evolution and Networks in Ancient and Widespread Symbioses between Mucoromycotina and Liverworts. Mycorrhiza 2019, 29, 551–565.
- Chaudhary, S.; Polaino, S.; Shakya, V.P.S.; Idnurm, A. A New Genetic Linkage Map of the Zygomycete Fungus Phycomyces blakesleeanus. PLoS ONE 2013, 8, e58931.
- Linde, J.; Schwartze, V.; Binder, U.; Lass-Flörl, C.; Voigt, K.; Horn, F. De Novo Whole-Genome Sequence and Genome Annotation of Lichtheimia ramosa. Genome Announc. 2014, 2, e00888-14.
- Schwartze, V.U.; Winter, S.; Shelest, E.; Marcet-Houben, M.; Horn, F.; Wehner, S.; Linde, J.; Valiante, V.; Sammeth, M.; Riege, K.; et al. Gene Expansion Shapes Genome Architecture in the Human Pathogen Lichtheimia corymbifera: An Evolutionary Genomics Analysis in the Ancient Terrestrial Mucorales (Mucoromycotina). PLoS Genet. 2014, 10, e1004496.
- Torres-Cruz, T.J.; Billingsley Tobias, T.L.; Almatruk, M.; Hesse, C.N.; Kuske, C.R.; Desirò, A.; Benucci, G.M.N.; Bonito, G.; Stajich, J.E.; Dunlap, C.; et al. Bifiguratus adelaidae, Gen. et Sp. Nov., a New Member of Mucoromycotina in Endophytic and Soil-Dwelling Habitats. Mycologia 2017, 109, 363–378.
- Hirose, D.; Degawa, Y.; Inaba, S.; Tokumasu, S. The Anamorphic Genus Calcarisporiella Is a New Member of the Mucoromycotina. Mycoscience 2012, 53, 256–260.
- Nagy, L.G.; Petkovits, T.; Kovács, G.M.; Voigt, K.; Vágvölgyi, C.; Papp, T. Where Is the Unseen Fungal Diversity Hidden? A Study of Mortierella Reveals a Large Contribution of Reference Collections to the Identification of Fungal Environmental Sequences. New Phytol. 2011, 191, 789–794.
- Vandepol, N.; Liber, J.; Desirò, A.; Na, H.; Kennedy, M.; Barry, K.; Grigoriev, I.V.; Miller, A.N.; O’Donnell, K.; Stajich, J.E.; et al. Resolving the Mortierellaceae Phylogeny through Synthesis of Multi-Gene Phylogenetics and Phylogenomics. Fungal Divers. 2020, 104, 267–289.
- Smith, M.E.; Gryganskyi, A.; Bonito, G.; Nouhra, E.; Moreno-Arroyo, B.; Benny, G. Phylogenetic Analysis of the Genus Modicella Reveals an Independent Evolutionary Origin of Sporocarp-Forming Fungi in the Mortierellales. Fungal Genet. Biol. 2013, 61, 61–68.
- Macias, A.M.; Geiser, D.M.; Stajich, J.E.; Łukasik, P.; Veloso, C.; Bublitz, D.C.; Berger, M.C.; Boyce, G.R.; Hodge, K.; Kasson, M.T. Evolutionary Relationships among Massospora Spp. (Entomophthorales), Obligate Pathogens of Cicadas. Mycologia 2020, 112, 1060–1074.
- Bonito, G.; Hameed, K.; Ventura, R.; Krishnan, J.; Schadt, C.W.; Vilgalys, R. Isolating a Functionally Relevant Guild of Fungi from the Root Microbiome of Populus. Fungal Ecol. 2016, 22, 35–42.
- Vandepol, N.; Liber, J.; Yocca, A.; Matlock, J.; Edger, P.; Bonito, G. Linnemannia elongata (Mortierellaceae) Stimulates Arabidopsis thaliana Aerial Growth and Responses to Auxin, Ethylene, and Reactive Oxygen Species. PLoS ONE 2022, 17, e0261908.
- Zhang, K.; Bonito, G.; Hsu, C.-M.; Hameed, K.; Vilgalys, R.; Liao, H.-L. Mortierella elongata Increases Plant Biomass among Non-Leguminous Crop Species. Agronomy 2020, 10, 754.
- Wurlitzer, J.M.; Stanišić, A.; Wasmuth, I.; Jungmann, S.; Fischer, D.; Kries, H.; Gressler, M. Bacterial-like Nonribosomal Peptide Synthetases Produce Cyclopeptides in the Zygomycetous Fungus Mortierella alpina. Appl. Environ. Microbiol. 2021, 87, e02051-20.
- Remy, W.; Taylor, T.N.; Hass, H.; Kerp, H. Four Hundred-Million-Year-Old Vesicular Arbuscular Mycorrhizae. Proc. Natl. Acad. Sci. USA 1994, 91, 11841–11843.
- Smith, S.; Read, D. Mycorrhizal Symbiosis, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008; ISBN 978-0-12-370526-6.
- Hijri, M.; Sanders, I.R. The Arbuscular Mycorrhizal Fungus Glomus intraradices Is Haploid and Has a Small Genome Size in the Lower Limit of Eukaryotes. Fungal Genet. Biol. 2004, 41, 253–261.
- Sędzielewska, K.A.; Fuchs, J.; Temsch, E.M.; Baronian, K.; Watzke, R.; Kunze, G. Estimation of the Glomus intraradices Nuclear DNA Content. New Phytol. 2011, 192, 794–797.
- Maeda, T.; Kobayashi, Y.; Kameoka, H.; Okuma, N.; Takeda, N.; Yamaguchi, K.; Bino, T.; Shigenobu, S.; Kawaguchi, M. Evidence of Non-Tandemly Repeated RDNAs and Their Intragenomic Heterogeneity in Rhizophagus irregularis. Commun. Biol. 2018, 1, 87.
- Venice, F.; Ghignone, S.; Salvioli di Fossalunga, A.; Amselem, J.; Novero, M.; Xianan, X.; Sędzielewska Toro, K.; Morin, E.; Lipzen, A.; Grigoriev, I.V.; et al. At the Nexus of Three Kingdoms: The Genome of the Mycorrhizal Fungus Gigaspora margarita Provides Insights into Plant, Endobacterial and Fungal Interactions. Environ. Microbiol. 2020, 22, 122–141.
- Ropars, J.; Toro, K.S.; Noel, J.; Pelin, A.; Charron, P.; Farinelli, L.; Marton, T.; Krüger, M.; Fuchs, J.; Brachmann, A.; et al. Evidence for the Sexual Origin of Heterokaryosis in Arbuscular Mycorrhizal Fungi. Nat. Microbiol. 2016, 1, 16033.
- Idnurm, A.; Walton, F.J.; Floyd, A.; Heitman, J. Identification of the Sex Genes in an Early Diverged Fungus. Nature 2008, 451, 193–196.
- Kuhn, G.; Hijri, M.; Sanders, I.R. Evidence for the Evolution of Multiple Genomes in Arbuscular Mycorrhizal Fungi. Nature 2001, 414, 745–748.
- Pawlowska, T.E.; Taylor, J.W. Organization of Genetic Variation in Individuals of Arbuscular Mycorrhizal Fungi. Nature 2004, 427, 733–737.
- Wewer, V.; Brands, M.; Dörmann, P. Fatty Acid Synthesis and Lipid Metabolism in the Obligate Biotrophic Fungus Rhizophagus irregularis during Mycorrhization of Lotus japonicus. Plant J. 2014, 79, 398–412.
- Tang, N.; San Clemente, H.; Roy, S.; Bécard, G.; Zhao, B.; Roux, C. A Survey of the Gene Repertoire of Gigaspora rosea Unravels Conserved Features among Glomeromycota for Obligate Biotrophy. Front. Microbiol. 2016, 7, 233.
- Kobayashi, Y.; Maeda, T.; Yamaguchi, K.; Kameoka, H.; Tanaka, S.; Ezawa, T.; Shigenobu, S.; Kawaguchi, M. The Genome of Rhizophagus clarus HR1 Reveals a Common Genetic Basis for Auxotrophy among Arbuscular Mycorrhizal Fungi. BMC Genom. 2018, 19, 465.
- Bravo, A.; Brands, M.; Wewer, V.; Dörmann, P.; Harrison, M.J. Arbuscular Mycorrhiza-Specific Enzymes FatM and RAM2 Fine-Tune Lipid Biosynthesis to Promote Development of Arbuscular Mycorrhiza. New Phytol. 2017, 214, 1631–1645.
- Jiang, Y.; Wang, W.; Xie, Q.; Liu, N.; Liu, L.; Wang, D.; Zhang, X.; Yang, C.; Chen, X.; Tang, D.; et al. Plants Transfer Lipids to Sustain Colonization by Mutualistic Mycorrhizal and Parasitic Fungi. Science 2017, 356, 1172–1175.
- Keymer, A.; Pimprikar, P.; Wewer, V.; Huber, C.; Brands, M.; Bucerius, S.L.; Delaux, P.-M.; Klingl, V.; von Röpenack-Lahaye, E.; Wang, T.L.; et al. Lipid Transfer from Plants to Arbuscular Mycorrhiza Fungi. eLife 2017, 6, e29107.
- Luginbuehl, L.H.; Menard, G.N.; Kurup, S.; Van Erp, H.; Radhakrishnan, G.V.; Breakspear, A.; Oldroyd, G.E.D.; Eastmond, P.J. Fatty Acids in Arbuscular Mycorrhizal Fungi Are Synthesized by the Host Plant. Science 2017, 356, 1175–1178.
- Kameoka, H.; Tsutsui, I.; Saito, K.; Kikuchi, Y.; Handa, Y.; Ezawa, T.; Hayashi, H.; Kawaguchi, M.; Akiyama, K. Stimulation of Asymbiotic Sporulation in Arbuscular Mycorrhizal Fungi by Fatty Acids. Nat. Microbiol. 2019, 4, 1654–1660.
- Sugiura, Y.; Akiyama, R.; Tanaka, S.; Yano, K.; Kameoka, H.; Marui, S.; Saito, M.; Kawaguchi, M.; Akiyama, K.; Saito, K. Myristate Can Be Used as a Carbon and Energy Source for the Asymbiotic Growth of Arbuscular Mycorrhizal Fungi. Proc. Natl. Acad. Sci. USA 2020, 117, 25779–25788.
- Sperschneider, J.; Yildirir, G.; Rizzi, Y.; Malar, C.M.; Sorwar, E.; Chen, E.C.; Iwasaki, W.; Brauer, E.K.; Bosnich, W.; Gutjahr, C.; et al. Resolving the Haplotypes of Arbuscular Mycorrhizal Fungi Highlights the Role of Two Nuclear Populations in Host Interactions. bioRxiv 2023.
- Hughes, D.P.; Araújo, J.P.M.; Loreto, R.G.; Quevillon, L.; de Bekker, C.; Evans, H.C. From So Simple a Beginning: The Evolution of Behavioral Manipulation by Fungi. Adv. Genet. 2016, 94, 437–469.
- Reynolds, N.K.; Stajich, J.E.; Benny, G.L.; Barry, K.; Mondo, S.; LaButti, K.; Lipzen, A.; Daum, C.; Grigoriev, I.V.; Ho, H.-M.; et al. Mycoparasites, Gut Dwellers, and Saprotrophs: Phylogenomic Reconstructions and Comparative Analyses of Kickxellomycotina Fungi. Genome Biol. Evol. 2023, 15, evac185.
- Davis, W.J.; Amses, K.R.; Benny, G.L.; Carter-House, D.; Chang, Y.; Grigoriev, I.; Smith, M.E.; Spatafora, J.W.; Stajich, J.E.; James, T.Y. Genome-Scale Phylogenetics Reveals a Monophyletic Zoopagales (Zoopagomycota, Fungi). Mol. Phylogenetics Evol. 2019, 133, 152–163.
- Vilela, R.; Mendoza, L. Human Pathogenic Entomophthorales. Clin. Microbiol. Rev. 2018, 31, e00014-18.
- Lazarus, K.L.; Benny, G.L.; Ho, H.-M.; Smith, M.E. Phylogenetic Systematics of Syncephalis (Zoopagales, Zoopagomycotina), a Genus of Ubiquitous Mycoparasites. Mycologia 2017, 109, 333–349.
- Lovett, B.; Macias, A.; Stajich, J.E.; Cooley, J.; Eilenberg, J.; de Fine Licht, H.H.; Kasson, M.T. Behavioral Betrayal: How Select Fungal Parasites Enlist Living Insects to Do Their Bidding. PLoS Pathog. 2020, 16, e1008598.
- Ahrendt, S.R.; Quandt, C.A.; Ciobanu, D.; Clum, A.; Salamov, A.; Andreopoulos, B.; Cheng, J.-F.; Woyke, T.; Pelin, A.; Henrissat, B.; et al. Leveraging Single-Cell Genomics to Expand the Fungal Tree of Life. Nat. Microbiol. 2018, 3, 1417–1428.
- Chang, Y.; Wang, S.; Sekimoto, S.; Aerts, A.L.; Choi, C.; Clum, A.; LaButti, K.M.; Lindquist, E.A.; Yee Ngan, C.; Ohm, R.A.; et al. Phylogenomic Analyses Indicate That Early Fungi Evolved Digesting Cell Walls of Algal Ancestors of Land Plants. Genome Biol. Evol. 2015, 7, 1590–1601.
- Wang, Y.; White, M.M.; Kvist, S.; Moncalvo, J.-M. Genome-Wide Survey of Gut Fungi (Harpellales) Reveals the First Horizontally Transferred Ubiquitin Gene from a Mosquito Host. Mol. Biol. Evol. 2016, 33, 2544–2554.
- Wang, Y.; Stata, M.; Wang, W.; Stajich, J.E.; White, M.M.; Moncalvo, J.-M. Comparative Genomics Reveals the Core Gene Toolbox for the Fungus-Insect Symbiosis. mBio 2018, 9, e00636-18.
- Tabima, J.F.; Trautman, I.A.; Chang, Y.; Wang, Y.; Mondo, S.; Kuo, A.; Salamov, A.; Grigoriev, I.V.; Stajich, J.E.; Spatafora, J.W. Phylogenomic Analyses of Non-Dikarya Fungi Supports Horizontal Gene Transfer Driving Diversification of Secondary Metabolism in the Amphibian Gastrointestinal Symbiont, Basidiobolus. G3 (Bethesda) 2020, 10, 3417–3433.
- Benny, G.L.; Smith, M.E.; Kirk, P.M.; Tretter, E.D.; White, M.M. Challenges and Future Perspectives in the Systematics of Kickxellomycotina, Mortierellomycotina, Mucoromycotina, and Zoopagomycotina. In Biology of Microfungi; Li, D.-W., Ed.; Fungal Biology; Springer International Publishing: Cham, Switzerland, 2016; pp. 65–126. ISBN 978-3-319-29137-6.
- Chuang, S.-C.; Ho, H.-M.; Reynolds, N.; Smith, M.E.; Benny, G.L.; Chien, C.-Y.; Tsai, J.-L. Preliminary Phylogeny of Coemansia (Kickxellales), with Descriptions of Four New Species from Taiwan. Mycologia 2017, 109, 815–831.
- Wang, Y.; White, M.M.; Moncalvo, J.-M. Draft Genome Sequence of Capniomyces stellatus, the Obligate Gut Fungal Symbiont of Stonefly. Genome Announc. 2016, 4, e00761-16.
- Tretter, E.D.; Johnson, E.M.; Benny, G.L.; Lichtwardt, R.W.; Wang, Y.; Kandel, P.; Novak, S.J.; Smith, J.F.; White, M.M. An Eight-Gene Molecular Phylogeny of the Kickxellomycotina, Including the First Phylogenetic Placement of Asellariales. Mycologia 2014, 106, 912–935.
- Lichtward, R.W. The Trichomycetes, Fungal Associates of Arthropods, 2nd ed.; Springer Science & Business Media: New York, NY, USA; Berlin/Heidelberg, Germany; Tokio, Japan, 2012.
- Wang, Y.; White, M.M.; Moncalvo, J.-M. Diversification of the Gut Fungi Smittium and Allies (Harpellales) Co-Occurred with the Origin of Complete Metamorphosis of Their Symbiotic Insect Hosts (Lower Diptera). Mol. Phylogenet. Evol. 2019, 139, 106550.
- White, M.M.; Guàrdia Valle, L.; Lichtwardt, R.W.; Siri, A.; Strongman, D.B.; William, R.T.; Gause, W.J.; Tretter, E.D. New Species and Emendations of Orphella: Taxonomic and Phylogenetic Reassessment of the Genus to Establish the Orphellales, for Stonefly Gut Fungi with a Twist. Mycologia 2018, 110, 147–178.
- Wang, Y.; Tretter, E.D.; Lichtwardt, R.W.; White, M.M. Overview of 75 Years of Smittium Research, Establishing a New Genus for Smittium culisetae, and Prospects for Future Revisions of the “Smittium” Clade. Mycologia 2013, 105, 90–111.
- White, M.M.; Siri, A.; Lichtwardt, R.W. Trichomycete Insect Symbionts in Great Smoky Mountains National Park and Vicinity. Mycologia 2006, 98, 333–352.
- Mariotti, M.; Salinas, G.; Gabaldón, T.; Gladyshev, V.N. Utilization of Selenocysteine in Early-Branching Fungal Phyla. Nat. Microbiol. 2019, 4, 759–765.
- Nie, Y.; Yu, D.-S.; Wang, C.-F.; Liu, X.-Y.; Huang, B. A Taxonomic Revision of the Genus Conidiobolus (Ancylistaceae, Entomophthorales): Four Clades Including Three New Genera. MycoKeys 2020, 66, 55–81.
- Humber, R.A. Synopsis of a Revised Classification for the Entomophthorales (Zygomycotina). Mycotaxon 1989, 34, 441–460.
- Nagahama, T.; Sato, H.; Shimazu, M.; Sugiyama, J. Phylogenetic Divergence of the Entomophthoralean Fungi: Evidence from Nuclear 18S Ribosomal RNA Gene Sequences. Mycologia 1995, 87, 203–209.
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A Genome-Scale Phylogeny of the Kingdom Fungi. Curr. Biol. 2021, 31, 1653–1665.e5.
More
Information
Subjects:
Mycology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
28 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No