Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Xiuling Cao | -- | 3075 | 2023-07-20 09:51:50 | | | |
| 2 | Dean Liu | -15 word(s) | 3060 | 2023-07-21 02:37:46 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Xiang, L.; Wang, Y.; Liu, S.; Liu, B.; Jin, X.; Cao, X. Targeting Protein Aggregates with Natural Products. Encyclopedia. Available online: https://encyclopedia.pub/entry/47019 (accessed on 26 July 2026).
Xiang L, Wang Y, Liu S, Liu B, Jin X, Cao X. Targeting Protein Aggregates with Natural Products. Encyclopedia. Available at: https://encyclopedia.pub/entry/47019. Accessed July 26, 2026.
Xiang, Lingzhi, Yanan Wang, Shenkui Liu, Beidong Liu, Xuejiao Jin, Xiuling Cao. "Targeting Protein Aggregates with Natural Products" Encyclopedia, https://encyclopedia.pub/entry/47019 (accessed July 26, 2026).
Xiang, L., Wang, Y., Liu, S., Liu, B., Jin, X., & Cao, X. (2023, July 20). Targeting Protein Aggregates with Natural Products. In Encyclopedia. https://encyclopedia.pub/entry/47019
Xiang, Lingzhi, et al. "Targeting Protein Aggregates with Natural Products." Encyclopedia. Web. 20 July, 2023.
Copy Citation
Protein aggregation is one of the hallmarks of aging and aging-related diseases, especially for the neurodegenerative diseases (NDs) such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic lateral sclerosis (ALS), and others.
natural products
neurodegenerative diseases
protein aggregation
Aβ
1. Introduction
Neurodegenerative diseases (NDs) are a heterogeneous group of disorders characterized by abnormal protein aggregation leading to the structural and functional degeneration of the central and peripheral nervous systems [1]. These diseases cause a large number of deaths and enormous medical costs worldwide, placing a heavy burden on patients, their families and society. According to the Global Alzheimer’s Disease Report, there are already more than 55 million people with Alzheimer’s disease (AD) worldwide, and this number is expected to rise to 78 million by 2030 and 152 million by 2050 [2]. Parkinson’s disease (PD) is the second most common and fastest growing ND in the world, with a global prevalence of more than 6 million people and a 2.5-fold increase from the previous generation of patients, and is expected to double again to more than 12 million by 2040 [3]. There is growing evidence that men are twice as likely as women to develop Parkinson’s disease, but women have higher mortality rates and a more rapid disease progression, placing an enormous burden on the population [4]. Huntington’s disease (HD) cases are distributed worldwide, with a prevalence of 4–10 per 100,000 in Western countries and approximately 5 per million in Asian populations [5][6]. The incidence of amyotrophic lateral sclerosis varies from country to country and region to region, from about (2–3/100,000) in Europe to about (0.7–0.8/100,000) in Asia, with huge annual treatment costs [7]. The common symptoms of these diseases are memory and cognitive impairment, as well as difficulties with speech and movement, and they tend to be more common in older people [8].
In these diseases, one or more different pathologically aggregation-prone polypeptides misfold and are packaged into large insoluble inclusion bodies. For example, the two hallmark pathological the features of AD are extracellular amyloid plaques composed of Aβ peptides and intracellular neurofibrillary tangles composed of hyperphosphorylated microtubule-associated protein tau [9][10]. PD is a movement disorder characterized by the accumulation of Lewy bodies in neurons, which are mainly composed of 𝛼-synuclein protein aggregates [11][12]. Due to CAG repeat expansion in the huntingtin (HTT) gene, the mutant Htt protein accumulates in neurons and forms deposits that produce cytotoxicity, leading to the development of HD [13]. And one of the reasons why ALS occurs is because the abnormally aggregated FUS protein state is more solidified, impairing its normal physiological function [14]. These diseases not only hinder people’s normal physical activities and increase their psychological stress, but also place a huge burden on society. However, there are no symptomatic drugs for these diseases. Therefore, finding ways to make the diseases more treatable has become a priority.
Natural products are a class of compounds isolated from plants or fungi that are biologically active and have a rich history of medicinal use [15]. In recent years, the biological activity, nutritional value, and potential health and therapeutic benefits of natural products have been intensively explored and studied [16][17]. Due to their neuroprotective effects, a variety of compounds from different sources have been proposed to have therapeutic efficacy in treating neurodegenerative diseases, and not only in alleviating their superficial symptoms [18][19][20]. Specifically, natural products can inhibit the formation of pathogenic protein aggregates and attenuate the neurotoxicity of pathogenic protein aggregates [16]. For example, natural products respond to autophagic pathways to reduce neurological damage such as oxidative stress from pathogenic protein aggregates [21][22][23]. In addition, natural products cleave β-amyloid structures to reduce aggregates formed by pathogenic proteins [24][25]. Similarly, natural products reduce levels of key enzyme activity that forms aggregates to inhibit oligomer formation [21][26][27]. Natural products offer new avenues for research into inhibiting the formation of disease-causing protein aggregates and thereby alleviating disease symptoms.
2. Natural Products Reduce Tau Aggregation by Affecting Aggregate Formation, Disaggregation, and Key Enzyme Activity
Tau is a phosphoprotein with a natively unfolded conformation that functions to stabilize microtubules in axons. Microtubules form the cytoskeleton of the cell and are essential for maintaining the structural integrity of the cell and transporting nutrients from the soma down the axon to the synaptic terminal [28][29][30][31]. In the adult human central nervous system, tau proteins exist as six heterodimers containing 0, 1, or 2 amino-terminal inserts and 3 or 4 microtubule-binding repeat (3R or 4R) domains. Those repeats, containing 31 or 32 amino acid residues, form domains that stabilize microtubules and promote microtubule assembly [32][33]. The ability of tau to bind to microtubules is also regulated by the post-translational modification of proteins, including phosphorylation, glycosylation, glycation, ubiquitination, sumoylation, and nitration [30][34]. Tau has multiple kinase phosphorylation sites and its functions are partly regulated by its phosphorylation status [35][36]. In all neurodegenerative diseases associated with the tau protein, this protein is present in a hyperphosphorylated form, which is responsible for its aggregation and leads to neuronal dysfunction and death. The aggregation of tau is a multi-step process; the initial step is the formation of the β-sheets of tau, i.e., MTBR regions of tau stacked on top of each other [37]. Then, it forms dimers and trimers, followed by small soluble oligomers. These small soluble oligomers form twisted tau filaments called PHFs, which subsequently form neurofibrillary tangles (NFTs) [38]. Tau-associated diseases are known as tauopathies and include Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), Pick’s disease, frontotemporal dementia (FTD), corticobasal degeneration, and variants of Parkinson’s disease (PD) and Lewy body dementia (LBD), for which NFTs are a common histopathological marker [34][39][40]. In addition, tau oligomers exhibit toxic effects in tauopathies prior to the formation of NFTs and are capable of potentiating neuronal damage, leading to neurodegeneration and traumatic brain injury [41]. Current drug strategies targeting the tau protein can be summarized as an inhibition of tau aggregation, inhibition of tau phosphorylation, reduction in tau levels and tau immunization [42].
In mouse cortical neuronal cells expressing induced wild-type tau and in primary cortical neurons, Fistein significantly reduced phosphorylated tau levels, which was highly dependent on TFEB and Nrf2 activation and occurred via selective autophagy by its cargo receptors [43]. Tau K18 is a widely used model for full-length tau proteins, as they exhibit very similar physiological and pathological functions [44]. In vitro, Fisetin has been shown to limit the extent of tau K18-protofibril formation by inhibiting tau K18 aggregation (Figure 1), resulting in shorter and thinner tau protofibrils. In HEK293/tau441, treatment with Fisetin reduced tau oligomers and significantly decreased the ratio of insoluble-to-soluble tau protein [45]. And treatment with Fisetin (20 mg/kg, i.p., 2 weeks) significantly reduced p-tau levels at Ser413 induced by Aβ(1–42) injection (i.c.v.) in the hippocampus of mice [46].
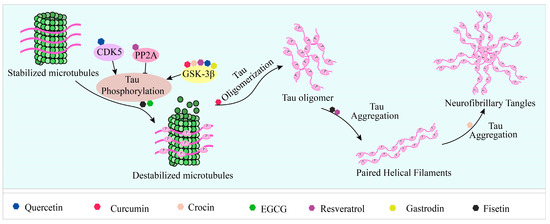
Figure 1. Natural products reduce tau aggregation, inhibit hyperphosphorylation, or act on formation processes. During the multistep process of tau aggregation, fisetin and EGCG reduce the phosphorylation level of tau. Curcumin, crocin, gastrodin, quercetin, and resveratrol can inhibit GSK3β activity and thus tau hyperphosphorylation. In addition, resveratrol and quercetin can inhibit tau phosphorylation by activating PP2A and inhibiting CDK5 activity, respectively. During the formation of tau oligomers, curcumin can inhibit its oligomerization. Fisetin and resveratrol can inhibit the accumulation of oligomers into PHFs, and curcumin can inhibit the further formation of NFTs from PHFs.
Crocin has been shown to inhibit neuronal death [47][48], protect rats from brain ischemia/reperfusion injury, and enhance long-term potentiation, learning, recognition and memory [49][50][51]. In vitro, crocin inhibited the conversion of tau protein into more aggregated conformations during the fibrillation process by binding to its intermediate structures and inhibited 50% of tau aggregates at a dose of 100 µg/mL [52]. In rats, co-treatment with 25 mg/kg crocin significantly reversed the level of acrolein-induced phosphorylation of tau in the cerebral cortex by attenuating the active forms of ERK and JNK kinases [53]. GSK-3β is the most important protein kinase that regulates tau phosphorylation, when it is overactivated, tau is hyperphosphorylated. In PC12-htau cells, tau is hyperphosphorylated at Thr231 and Ser199/Ser202 compared to PC12 cells. In PC12-htau it has been shown that trans-Crocin 4 decreases the amount and phosphorylation of tau at the pThr231 and pSer199/Ser202 epitopes, and inhibits the active forms of GSK3β and ERK1/2 kinases [54].
Resveratrol (RES) has pharmacological properties with antioxidant, anti-inflammatory, hepatoprotective, anti-diabetic, and anti-tumor effects [55][56]. It is believed to have therapeutic potential in the treatment of neurodegenerative diseases. For example, treatment with RES reduced tau phosphorylation in the hippocampus of diabetic mice fed a high-fat diet, resulting in improved memory impairment [57]. An in vitro ThT fluorescence assay showed that RES inhibited tau aggregation, resulting in the formation of smaller aggregates rather than long fibers. Moreover, it prevented extracellular tau oligomers from binding to N2a cells, reduced tau propagation, and decreased the levels of phosphorylated tau and tau oligomers in the brains of PS19 mice [58]. In addition to GSK-3β, calmodulin-dependent protein kinase II (CaMKII) and phosphoserine/phosphothreonine protein phosphatase-2A (PP2A) are also important enzymes involved in the regulation of tau protein hyperphosphorylation [59][60][61][62][63]. RES inhibited formaldehyde-induced increasing phosphorylation of GSK-3β and CaMKII protein levels to prevent tau protein hyperphosphorylation, thereby protecting N2a cells from formaldehyde-induced damage [64]. PP2A dephosphorylates tau, preventing its microtubule dissociation and PHF formation. MID1 is a negative regulator of PP2A and mediates the ubiquitin-specific degradation of PP2A. The loss of its function results in increased PP2A protein levels and activity [65]. Both in vitro and in vivo, RES treatment destabilized the ubiquitin ligase MID1 and its mRNA, which directly interfered with the MID1–α4–PP2A degradation complex by decreasing MID1 protein expression, leading to an increase in microtubule-associated PP2A activity and the time- and dose- dependent dephosphorylation of tau [66]. Similarly, in the brain of CdCl2-treated rats, trans-resveratrol inhibited tau phosphorylation by activating PP2A and inhibiting GSK3β activity. In particular, the inhibition of GSK3β activity was mediated by AMPK-induced activation of the PI3K/Akt signaling pathway [67]. In addition, RES inhibited alum-induced tau hyperphosphorylation at the Ser396 site in rat hippocampal slices by decreasing ERK1/2 activation and increasing GSK-3β Ser9 phosphorylation [68]. In optic nerve head astrocytes (ONHAs) undergoing oxidative stress, pretreatment with resveratrol not only increased cell viability, but also reduced the levels of activated caspases and dephosphorylation of the tau protein at Ser422, thereby reducing caspase-mediated tau cleavage and neurogenic fiber tangle (NFT) formation [69].
As mentioned above, quercetin, curcumin, and EGCG exerted potent neuroprotective effects in inhibiting Aβ formation and attenuating Aβ toxicity. In addition to this, they also show protective effects in terms of lowering tau phosphorylation levels and reducing the levels of aggregated tau. Quercetin-3-O-glucuronide (Q3G), a major quercetin metabolite in human plasma, has been reported to have potential neuroprotective effects [70]. Pretreatment with 10 µM quercetin or Q3G inhibited okadaic acid (OA)-induced phosphorylation of the tau protein in SH-SY5Y. An oral administration of quercetin also effectively attenuated overexpression of the tau protein phosphorylation in the hippocampus of mice during HFD feeding. Further experiments demonstrated that this was due to the activation of AMPK and inhibition of GSK3β activation by enhancing phosphorylation at the Ser 9 residue [71]. Cell cycle protein-dependent kinase 5 (CDK5) is one of the kinases that affect tau phosphorylation, and overactivated CDK5 activity leads to an abnormal phosphorylation of tau [72]. Quercetin inhibited CDK5 activity, blocked the Ca2+–calpain–p25–CDK5 signaling pathway, and inhibited tau phosphorylation at four sites (Ser396, Ser199, Thr205, and Thr231), thus exhibiting significant neuroprotective effects on OA-induced Ht22 cells [73]. In vitro, quercetin was shown through ThT fluorometry to inhibit tau fibrillization and disassemble pre-formed aggregates of the tau protein [74]. Curcumin has long been shown to inhibit GSK-3β activity and prevent tau hyperphosphorylation, thereby protecting SH-SY5Y from Aβ-induced mitochondrial dysfunction [75][76]. In vitro, curcumin has been shown to inhibit the formation of tau β-sheets, inhibit tau fibrillation, and degrade formed tau filaments, thereby reducing the level of aggregated tau, with 20 µM curcumin leading to 75 ± 10% disaggregation of tau aggregates [77]. As for EGCG, in vitro, it blocked K18ΔK280 aggregation and inhibited the formation of potentially proteotoxic oligomeric tau species [78]. In primary neurons, phospho-tau (p-S396/404, p-S262, and p-T231) and total tau levels decreased after 24 h of 50 µM EGCG treatment, but mRNA levels of tau were not affected. This suggests that the reduction in tau was due to clearance rather than transcriptional repression [79]. Other studies have also shown that EGCG binds tau in its phosphorylation region with an affinity of the same order of magnitude as kinases (0.5 mM), preventing it from contacting the protein and thus playing a key role in preventing tau aggregation [80].
In addition to the compounds listed above, there are also many potential therapeutic agents in tauopathies. Gastrodin reduced tau phosphorylation levels of Ser396, Ser199, and Thr231, and inhibited GSK3β kinase activity levels in the brains of APP/PS1 transgenic mice [81]. Morin, a natural bioflavonoid, reduces tau hyperphosphorylation by inhibiting GSK3β activity and the CDK5 signaling pathway in mice [82][83]. The monoterpene 1,8-cineole (CIN), present in many plant essential oils, attenuated the abnormal phosphorylation levels of the tau protein at the thr205, thr181, and ser396 sites induced by AGEs in vitro and in vivo [84]. Macelignan, a sort of lignan derived from Myristica fragrans mace, reduced tau phosphorylation in tau-overexpressing cells and primary neurons of 3× AD-transgene mice. It also promoted PP2A activity in tau-overexpressing cells [85]. In addition, plant-derived nobiletin, beta boswellic acid, huperzine A, and caffeine exhibited the inhibition of tau hyperphosphorylation in different mouse models, respectively [86][87][88][89]. Isobavachalcone is the main component extracted from Psoralea corylifolia. In vitro, isobavachalcone can inhibit heparin-induced tau K18 aggregation and break down mature fibrils into shorter and smaller fibrils or short fragments. Furthermore, in N2a cells, it reduced the proportion of apoptosis caused by phosphatidylserine-induced tau K18 oligomer, from 40% to 10%. It also reduced the level of tau phosphorylation by regulating the levels of GSK3β and PP2A [90]. Limonoids (nimbin and salannin), isolated from neem fruit, were able to inhibit hTau40w aggregation and instead form thin, short, fragile tau fragments [91].
3. Natural Products Inhibit, Degrade, and Remodel α-Syn Fibrils to Reduce Accumulation and Toxicity
Alpha-synuclein (α-Syn) is an intrinsically disordered protein [92] that is abundant in the central nervous system [93] and transforms into cross-β-sheets rich amyloid by self-assembly under physiological conditions via partially folded intermediates and soluble oligomers [94]. Some aggregated species of α-Syn formed along the fibrillation are highly toxic and capable of interfering with the functions of different organelles such as mitochondria, endoplasmic reticulum, and plasma membrane [95][96][97]. Furthermore, it may increase oxidative stress, causing severe damages in dopaminergic cells [98][99]. Therefore, molecules that inhibit α-synuclein fibrillization and stabilize it in a non-toxic state can serve as therapeutic molecules that both prevent the accumulation of aggregated α-syn and maintain normal physiological concentrations of α-syn [100].
Studies have identified small molecules, nanoparticles, peptides, and polymers that have the ability to inhibit α-synuclein fibril formation or destabilize preformed α-syn fibrils (Figure 2). Curcumin has been mentioned above for its significant inhibitory effect on the formation of aggregates of Aβ and tau [101][102][103][104]. Curcumin has also been shown to inhibit the aggregation of α-syn in vitro and attenuate the toxicity of α-syn oligomers in cells [105][106]. In addition, curcumin prevented lipopolysaccharide-induced increases in α-syn gene expression in rats [107]. Due to the instability of curcumin in solution, stable curcumin analogues have raised some concerns. Curcumin pyrazole and its derivative (N-(3-nitrophenylpyrazole) curcumin inhibited the aggregation, protofibrosis, and toxicity of α-syn. Through biochemical, biophysical, and cell-based assays, both have been found to exhibit significant efficacy not only in arresting fibrillization and destroying pre-formed fibrils, but also in preventing formation of the A11 conformation in proteins, which can have toxic effects [108]. EGCG is another natural product that has received particular attention for targeting α-syn fibrillization due to its high availability and low toxicity [109][110]. In vitro, EGCG effectively inhibited α-syn fibrillogenesis by binding to naturally unstructured α-syn monomers and preventing their conversion into stable, β-sheet-rich structures. Instead, it promoted the formation of a novel non-structural, non-toxic α-synuclein [111]. In the rat immortalized oligodendrocyte cell line, OLN-93, EGCG immobilized the C-terminal region, moderately reduced the degree of oligomer binding to the membrane, and inhibited the ability of pre-formed oligomers to permeabilize vesicles and induce cytotoxicity [112]. ‘Active’ oligomers (AOs), characterized as a meta-stable and β-sheet-free species, exhibit rapid self-assembly into the radiating amyloid fibrils (RAFs) on the liposome surface, leading to drastic disruption of the membrane structures [113]. EGCG suppressed the membrane-disrupting radiating amyloid fibril formation on the surface of liposomal membranes, thus protecting the cells that can be readily affected by Aos [114]. According to the results of a molecular dynamics simulation, EGCG can disrupt the β-sheet structure and reduce the β-sheet content to remodel α-syn fibrils [115][116].
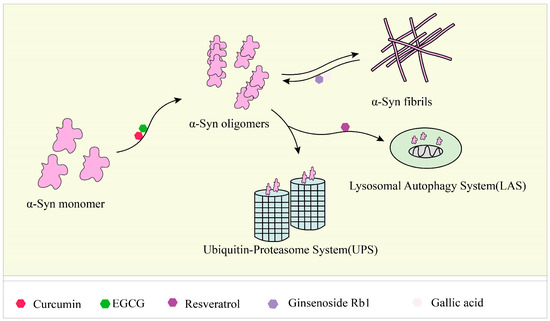
Figure 2. Responses of natural products to α-syn fibrils: inhibition, degradation, and remodeling. In the fibrosis of α-syn, curcumin and EGCG can inhibit its conversion from a monomer to an oligomer, and ginsenoside Rb1 and gallic acid can degrade the formed fibrils. Resveratrol can induce the autophagic degradation of α-syn.
There is evidence that alterations in the autophagy lysosomal pathway of α-synuclein degradation may be preferentially involved in neuronal death and contribute to the pathogenesis of PD [117][118]. RES-activated SIRT1, deacetylated microtubule-associated protein 1 light chain 3 (LC3), and caused the autophagic degradation of α-syn in dopaminergic neurons [119]. Studies have shown that ginsenoside Rb1 effectively inhibited α-syn fibrillation, with an inhibition rate of approximately 90% at 25 µM and incubation for two days. Additionally, Rb1 exhibited a strong ability to decompose preformed fibrils and inhibit the seeded polymerization of α-syn [120]. In vitro thioflavin T fluorescence assays and transmission electron microscopy imaging results showed that GA can inhibit the formation of amyloid fibrils by α-syn and disaggregate preformed α-syn amyloid fibrils. For soluble non-toxic oligomers without β-sheet content, GA can bind to them to stabilize their structure [119][121]. Triptolide (T10) is a monomeric compound isolated from Tripterygium wilfordii Hook f (TWHF). It has anti-inflammatory and anti-tumor activities, as well as neuroprotective effects [122][123]. In neuronal cells, T10 decreased the expression level of α-syn and acted as an autophagy inducer to promote the degradation of α-syn without disturbing lysosomal function [124].
In addition to the above, other compounds have been found to have effects on α-syn aggregation in vitro. For example, the components of saffron, crocin-1, crocin-2, and crocetin, inhibited α-syn aggregation, and dissociated α-syn fibrils [50]. The compounds in Rose damascena can inhibit α-syn fibrillation and oligomer toxicity [125]. In addition, the combined action of the compounds offers a new possibility. Protocatechuic acid (PCA) and hydroxytyrosol (HT) were able to reduce α-syn toxicity. When PCA (100 μM) and HT (100 μM) were used in combination, they showed a higher inhibition of α-syn protofibril formation and destabilization of α-syn fibrils, of 88% and 62%, respectively [126].
References
- Sharma, N.; Tan, M.A.; An, S.S.A. Phytosterols: Potential metabolic modulators in neurodegenerative diseases. Int. J. Mol. Sci. 2021, 22, 12255.
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021 Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: Amsterdam, The Netherlands, 2021.
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinsons. Dis. 2018, 8, S3–S8.
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Parkinsons. Dis. 2019, 9, 501–515.
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091.
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750.
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226.
- Hassan, S.S.U.; Samanta, S.; Dash, R.; Karpinski, T.M.; Habibi, E.; Sadiq, A.; Ahmadi, A.; Bunagu, S. The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: Focus on the role of oxidative stress. Front. Pharmacol. 2022, 13, 1015835.
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890.
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Sci. N. Y. 1997, 276, 2045–2047.
- Bates, G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 2003, 361, 1642–1644.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Salem, M.A.; Perez de Souza, L.; Serag, A.; Fernie, A.R.; Farag, M.A.; Ezzat, S.M.; Alseekh, S. Metabolomics in the context of plant natural products research: From sample preparation to metabolite analysis. Metabolites 2020, 10, 37.
- Mohd Sairazi, N.S.; Sirajudeen, K.N.S. Natural products and their bioactive compounds: Neuroprotective potentials against neurodegenerative diseases. Evid. Based Complement. Alternat. Med. 2020, 2020, 6565396.
- Zhang, L.; Song, J.; Kong, L.; Yuan, T.; Li, W.; Zhang, W.; Hou, B.; Lu, Y.; Du, G. The strategies and techniques of drug discovery from natural products. Pharmacol. Therapeut. 2020, 216, 107686.
- Essa, M.M.; Vijayan, R.K.; Castellano-Gonzalez, G.; Memon, M.A.; Braidy, N.; Guillemin, G.J. Neuroprotective effect of natural products against Alzheimer’s disease. Neurochem. Res. 2012, 37, 1829–1842.
- Chen, X.; Drew, J.; Berney, W.; Lei, W. Neuroprotective natural products for Alzheimer’s disease. Cells 2021, 10, 1309.
- Wang, Z.; He, C.; Shi, J.S. Natural products for the treatment of neurodegenerative diseases. Curr. Med. Chem. 2020, 27, 5790–5828.
- Taylor, E.; Kim, Y.; Zhang, K.; Chau, L.; Nguyen, B.C.; Rayalam, S.; Wang, X. Antiaging Mechanism of natural compounds: Effects on autophagy and oxidative stress. Molecules 2022, 27, 4396.
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815.
- Cui, X.; Lin, Q.; Liang, Y. Plant-derived antioxidants protect the nervous system from aging by inhibiting oxidative stress. Front. Aging Neurosci. 2020, 12, 209.
- Lee, J.H.; Ahn, N.H.; Choi, S.B.; Kwon, Y.; Yang, S.H. Natural products targeting amyloid beta in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 2341.
- Witter, S.; Samoson, A.; Vilu, R.; Witter, R. Screening of nutraceuticals and plant extracts for inhibition of amyloid-β fibrillation. J. Alzheimers Dis. 2020, 73, 1003–1012.
- Noori, T.; Dehpour, A.R.; Sureda, A.; Sobarzo-Sanchez, E.; Shirooie, S. Role of natural products for the treatment of Alzheimer’s disease. Eur. J. Pharmacol. 2021, 898, 173974.
- Rocha-Gonzalez, H.I.; Ambriz-Tututi, M.; Granados-Soto, V. Resveratrol: A natural compound with pharmacological potential in neurodegenerative diseases. CNS Neurosci. Ther. 2008, 14, 234–247.
- Dani, M.; Brooks, D.J.; Edison, P. Tau imaging in neurodegenerative diseases. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1139–1150.
- Giacobini, E.; Gold, G. Alzheimer disease therapy--moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686.
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622.
- Lee, H.G.; Perry, G.; Moreira, P.I.; Garrett, M.R.; Liu, Q.; Zhu, X.; Takeda, A.; Nunomura, A.; Smith, M.A. Tau phosphorylation in Alzheimer’s disease: Pathogen or protector? Trends Mol. Med. 2005, 11, 164–169.
- Chabrier, M.A.; Cheng, D.; Castello, N.A.; Green, K.N.; LaFerla, F.M. Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease. Neurobiol. Dis. 2014, 64, 107–117.
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Bio. 1977, 116, 227–247.
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672.
- Herrup, K.; Carrillo, M.C.; Schenk, D.; Cacace, A.; Desanti, S.; Fremeau, R.; Bhat, R.; Glicksman, M.; May, P.; Swerdlow, R.; et al. Beyond amyloid: Getting real about nonamyloid targets in Alzheimer’s disease. Alzheimers Dement. 2013, 9, 452–458.e1.
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 2005, 1739, 280–297.
- von Bergen, M.; Barghorn, S.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta 2005, 1739, 158–166.
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713.
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Their relevance for understanding the neurogenerative process. Ann. N. Y. Acad. Sci. 2000, 920, 74–83.
- Hutton, M. Molecular genetics of chromosome 17 tauopathies. Ann. N. Y. Acad. Sci. 2000, 920, 63–73.
- Shafiei, S.S.; Guerrero-Munoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83.
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415.
- Kim, S.; Choi, K.J.; Cho, S.J.; Yun, S.M.; Jeon, J.P.; Koh, Y.H.; Song, J.; Johnson, G.V.; Jo, C. Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci. Rep. 2016, 6, 24933.
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678.
- Xiao, S.; Lu, Y.; Wu, Q.; Yang, J.; Chen, J.; Zhong, S.; Eliezer, D.; Tan, Q.; Wu, C. Fisetin inhibits tau aggregation by interacting with the protein and preventing the formation of beta-strands. Int. J. Biol. Macromol. 2021, 178, 381–393.
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice. Mol. Neurobiol. 2017, 54, 2269–2285.
- Soeda, S.; Ochiai, T.; Paopong, L.; Tanaka, H.; Shoyama, Y.; Shimeno, H. Crocin suppresses tumor necrosis factor-alpha-induced cell death of neuronally differentiated PC-12 cells. Life Sci. 2001, 69, 2887–2898.
- Ochiai, T.; Ohno, S.; Soeda, S.; Tanaka, H.; Shoyama, Y.; Shimeno, H. Crocin prevents the death of rat pheochromyctoma (PC-12) cells by its antioxidant effects stronger than those of alpha-tocopherol. Neurosci. Lett. 2004, 362, 61–64.
- Georgiadou, G.; Tarantilis, P.A.; Pitsikas, N. Effects of the active constituents of Crocus sativus L. crocins, in an animal model of obsessive-compulsive disorder. Neurosci. Lett. 2012, 528, 27–30.
- Inoue, E.; Shimizu, Y.; Masui, R.; Hayakawa, T.; Tsubonoya, T.; Hori, S.; Sudoh, K. Effects of saffron and its constituents, crocin-1, crocin-2, and crocetin on alpha-synuclein fibrils. J. Nat. Med. 2018, 72, 274–279.
- Sugiura, M.; Shoyama, Y.; Saito, H.; Abe, K. The effects of ethanol and crocin on the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Jap. J. Of. Pharmacol. 1995, 67, 395–397.
- Karakani, A.M.; Riazi, G.; Ghaffari, S.M.; Ahmadian, S.; Mokhtari, F.; Firuzi, M.J.; Bathaie, S.Z. Inhibitory effect of corcin on aggregation of 1N/4R human tau protein in vitro. Iran. J. Basic Med. Sci. 2015, 18, 485–492.
- Rashedinia, M.; Lari, P.; Abnous, K.; Hosseinzadeh, H. Protective effect of crocin on acrolein-induced tau phosphorylation in the rat brain. Acta Neurobiol. Exp. 2015, 75, 208–219.
- Chalatsa, I.; Arvanitis, D.A.; Koulakiotis, N.S.; Giagini, A.; Skaltsounis, A.L.; Papadopoulou-Daifoti, Z.; Tsarbopoulos, A.; Sanoudou, D. The crocus sativus compounds trans-crocin 4 and trans-crocetin modulate the amyloidogenic pathway and tau misprocessing in Alzheimer disease neuronal cell culture models. Front. Neurosci. 2019, 13, 249.
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food. Chem. 2002, 50, 3337–3340.
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506.
- Jeon, B.T.; Jeong, E.A.; Shin, H.J.; Lee, Y.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes 2012, 61, 1444–1454.
- Sun, X.Y.; Dong, Q.X.; Zhu, J.; Sun, X.; Zhang, L.F.; Qiu, M.; Yu, X.L.; Liu, R.T. Resveratrol rescues tau-induced cognitive deficits and neuropathology in a mouse model of tauopathy. Curr. Alzheimer Res. 2019, 16, 710–722.
- Gong, C.X.; Shaikh, S.; Wang, J.Z.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J. Neurochem. 1995, 65, 732–773.
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927.
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412.
- Wang, Y.J.; Chen, G.H.; Hu, X.Y.; Lu, Y.P.; Zhou, J.N.; Liu, R.Y. The expression of calcium/calmodulin-dependent protein kinase II-alpha in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res. 2005, 1031, 101–108.
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neurotaph. Exp. Neur. 1999, 58, 1010–1019.
- He, X.; Li, Z.; Rizak, J.D.; Wu, S.; Wang, Z.; He, R.; Su, M.; Qin, D.; Wang, J.; Hu, X. Resveratrol attenuates formaldehyde induced hyperphosphorylation of tau protein and cytotoxicity in N2a cells. Front. Neurosci. 2016, 10, 598.
- Trockenbacher, A.; Suckow, V.; Foerster, J.; Winter, J.; Krauss, S.; Ropers, H.H.; Schneider, R.; Schweiger, S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 2001, 29, 287–294.
- Schweiger, S.; Matthes, F.; Posey, K.; Kickstein, E.; Weber, S.; Hettich, M.M.; Pfurtscheller, S.; Ehninger, D.; Schneider, R.; Krauß, S. Resveratrol induces dephosphorylation of Tau by interfering with the MID1-PP2A complex. Sci. Rep. 2017, 7, 13753.
- Shati, A.A.; Alfaifi, M.Y. Trans-resveratrol Inhibits Tau phosphorylation in the brains of control and cadmium chloride-treated rats by activating PP2A and PI3K/Akt induced-inhibition of GSK3beta. Neurochem. Res. 2019, 44, 357–373.
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Resveratrol ameliorates tau hyperphosphorylation at Ser396 Site and oxidative damage in rat hippocampal slices exposed to vanadate: Implication of ERK1/2 and GSK-3beta signaling cascades. J. Agric. Food Chem. 2017, 65, 9626–9634.
- Means, J.C.; Lopez, A.A.; Koulen, P. Resveratrol protects optic nerve head astrocytes from oxidative stress-induced cell death by preventing caspase-3 activation, tau dephosphorylation at Ser(422) and formation of misfolded protein aggregates. Cell. Mol. Neurobiol. 2020, 40, 911–926.
- Ishisaka, A.; Mukai, R.; Terao, J.; Shibata, N.; Kawai, Y. Specific localization of quercetin-3-O-glucuronide in human brain. Arch. Biochem. Biophys. 2014, 557, 11–17.
- Chen, J.; Deng, X.; Liu, N.; Li, M.; Liu, B.; Fu, Q.; Qu, R.; Ma, S. Quercetin attenuates tau hyperphosphorylation and improves cognitive disorder via suppression of ER stress in a manner dependent on AMPK pathway. J. Funct. Foods 2016, 22, 463–476.
- Kimura, T.; Ishiguro, K.; Hisanaga, S. Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 2014, 7, 65.
- Shen, X.Y.; Luo, T.; Li, S.; Ting, O.Y.; He, F.; Xu, J.; Wang, H.Q. Quercetin inhibits okadaic acid-induced tau protein hyperphosphorylation through the Ca2+-calpain-p25-CDK5 pathway in HT22 cells. Int. J. Mol. Med. 2018, 41, 1138–1146.
- Kumar, S.; Krishnakumar, V.G.; Morya, V.; Gupta, S.; Datta, B. Nanobiocatalyst facilitated aglycosidic quercetin as a potent inhibitor of tau protein aggregation. Int. J. Biol. Macromol. 2019, 138, 168–180.
- Huang, H.C.; Xu, K.; Jiang, Z.F. Curcumin-mediated neuroprotection against amyloid-beta-induced mitochondrial dysfunction involves the inhibition of GSK-3beta. J. Alzheimers Dis. 2012, 32, 981–996.
- Huang, H.C.; Tang, D.; Xu, K.; Jiang, Z.F. Curcumin attenuates amyloid-beta-induced tau hyperphosphorylation in human neuroblastoma SH-SY5Y cells involving PTEN/Akt/GSK-3beta signaling pathway. J. Recept. Signal Transduct. Res. 2014, 34, 26–37.
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimers. Dis. 2017, 60, 999–1014.
- Wobst, H.J.; Sharma, A.; Diamond, M.I.; Wanker, E.E.; Bieschke, J. The green tea polyphenol (-)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS Lett. 2015, 589, 77–83.
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31.
- Gueroux, M.; Fleau, C.; Slozeck, M.; Laguerre, M.; Pianet, I. Epigallocatechin 3-gallate as an inhibitor of tau phosphorylation and aggregation: A molecular and structural insight. J. Prev. Alzheimers Dis. 2017, 4, 218–225.
- Zeng, Y.-Q.; Gu, J.-H.; Chen, L.; Zhang, T.-T.; Zhou, X.-F. Gastrodin as a multi-target protective compound reverses learning memory deficits and AD-like pathology in APP/PS1 transgenic mice. J. Funct. Foods 2021, 77, 104324.
- Gong, E.J.; Park, H.R.; Kim, M.E.; Piao, S.; Lee, E.; Jo, D.G.; Chung, H.Y.; Ha, N.C.; Mattson, M.P.; Lee, J. Morin attenuates tau hyperphosphorylation by inhibiting GSK3beta. Neurobiol. Dis. 2011, 44, 223–230.
- Du, Y.; Qu, J.; Zhang, W.; Bai, M.; Zhou, Q.; Zhang, Z.; Li, Z.; Miao, J. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology 2016, 108, 1–13.
- An, F.; Bai, Y.; Xuan, X.; Bian, M.; Zhang, G.; Wei, C. 1,8-Cineole ameliorates advanced glycation end products-induced Alzheimer’s disease-like pathology in vitro and in vivo. Molecules 2022, 27, 3913.
- Hitl, M.; Kladar, N.; Gavarić, N.; Božin, B. Rosmarinic acid-human pharmacokinetics and health benefits. Planta Medica 2021, 87, 273–282.
- Nakajima, A.; Aoyama, Y.; Nguyen, T.T.; Shin, E.J.; Kim, H.C.; Yamada, S.; Nakai, T.; Nagai, T.; Yokosuka, A.; Mimaki, Y.; et al. Nobiletin, a citrus flavonoid, ameliorates cognitive impairment, oxidative burden, and hyperphosphorylation of tau in senescence-accelerated mouse. Behav. Brain Res. 2013, 250, 351–360.
- Huang, X.T.; Qian, Z.M.; He, X.; Gong, Q.; Wu, K.C.; Jiang, L.R.; Lu, L.N.; Zhu, Z.J.; Zhang, H.Y.; Yung, W.H.; et al. Reducing iron in the brain: A novel pharmacologic mechanism of huperzine A in the treatment of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1045–1054.
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Muller, C.E.; Buee, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090.
- Shasaltaneh, M.D.; Naghdi, N.; Ramezani, S.; Alizadeh, L.; Riazi, G.H. Protection of beta boswellic acid against streptozotocin-induced Alzheimer’s model by reduction of tau phosphorylation level and enhancement of reelin expression. Planta Med. 2022, 88, 367–379.
- Xiao, S.; Wu, Q.; Yao, X.; Zhang, J.; Zhong, W.; Zhao, J.; Liu, Q.; Zhang, M. Inhibitory effects of isobavachalcone on tau protein aggregation, tau phosphorylation, and oligomeric tau-induced apoptosis. ACS Chem. Neurosci. 2021, 12, 123–132.
- Gorantla, N.V.; Das, R.; Mulani, F.A.; Thulasiram, H.V.; Chinnathambi, S. Neem derivatives inhibits tau aggregation. J. Alzheimers Dis. Rep. 2019, 3, 169–178.
- Khare, S.D.; Chinchilla, P.; Baum, J. Multifaceted interactions mediated by intrinsically disordered regions play key roles in alpha synuclein aggregation. Curr. Opin. Struc. Biol. 2023, 80, 102579.
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220.
- Uversky, V.N.; Lee, H.J.; Li, J.; Fink, A.L.; Lee, S.J. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J. Biol. Chem. 2001, 276, 43495–43498.
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149.
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156.
- Uversky, V.N. Hot, Hotter, and Hottest Trends in α-Synuclein Research. Curr. Protein. Pept. Sci. 2015, 16, 682–687.
- Büeler, H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 2009, 218, 235–246.
- Niranjan, R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: Focus on astrocytes. Mol. Neurobiol. 2014, 49, 28–38.
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem. Biol. 2004, 11, 1513–1521.
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharmacol. 2008, 75, 787–809.
- Cole, G.M.; Teter, B.; Frautschy, S.A. Neuroprotective effects of curcumin. Ad. Exp. Med. Biol. 2007, 595, 197–212.
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377.
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901.
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199.
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res. 2011, 63, 439–444.
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits alpha-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360.
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin Pyrazole and its derivative (N-(3-Nitrophenylpyrazole) Curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of Wild type and Mutant alpha-Synuclein. Sci. Rep. 2015, 5, 9862.
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821.
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094.
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566.
- Lorenzen, N.; Nielsen, S.B.; Yoshimura, Y.; Vad, B.S.; Andersen, C.B.; Betzer, C.; Kaspersen, J.D.; Christiansen, G.; Pedersen, J.S.; Jensen, P.H.; et al. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J. Biol. Chem. 2014, 289, 21299–21310.
- Lee, J.H.; Hong, C.S.; Lee, S.; Yang, J.E.; Park, Y.I.; Lee, D.; Hyeon, T.; Jung, S.; Paik, S.R. Radiating amyloid fibril formation on the surface of lipid membranes through unit-assembly of oligomeric species of α-synuclein. PLoS ONE 2012, 7, e47580.
- Yang, J.E.; Rhoo, K.Y.; Lee, S.; Lee, J.T.; Park, J.H.; Bhak, G.; Paik, S.R. EGCG-mediated protection of the membrane disruption and cytotoxicity caused by the ‘active oligomer’ of alpha-synuclein. Sci. Rep. 2017, 7, 17945.
- Liu, X.; Zhou, S.; Shi, D.; Bai, Q.; Liu, H.; Yao, X. Influence of EGCG on alpha-synuclein (alphaS) aggregation and identification of their possible binding mode: A computational study using molecular dynamics simulation. Chem. Biol. Drug Des. 2018, 91, 162–171.
- Yao, Y.; Tang, Y.; Wei, G. Epigallocatechin Gallate Destabilizes alpha-Synuclein Fibril by Disrupting the E46-K80 Salt-Bridge and Inter-protofibril Interface. ACS Chem. Neurosci. 2020, 11, 4351–4361.
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson's disease. Movement. Disorders. 2013, 28, 725–732.
- Bourdenx, M.; Bezard, E.; Dehay, B. Lysosomes and α-synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 2014, 8, 83.
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Menon, S.A.; Abul Khair, S.B.; Lu, J.H.; Safieh-Garabedian, B.; Al-Hayani, A.A.; Eliezer, D.; Li, M.; et al. Ginsenoside Rb1 inhibits fibrillation and toxicity of alpha-synuclein and disaggregates preformed fibrils. Neurobiol. Dis. 2015, 74, 89–101.
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic acid interacts with alpha-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta. 2014, 1844, 1481–1485.
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Abul Khair, S.B.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eliezer, D.; et al. Structure activity relationship of phenolic acid inhibitors of alpha-synuclein fibril formation and toxicity. Front. Aging. Neurosci. 2014, 6, 197.
- Gu, W.Z.; Chen, R.; Brandwein, S.; McAlpine, J.; Burres, N. Isolation, purification, and characterization of immunosuppressive compounds from tripterygium: Triptolide and tripdiolide. Int. J. Immunopharmaco. 1995, 17, 351–356.
- Zheng, Y.; Zhang, W.J.; Wang, X.M. Triptolide with potential medicinal value for diseases of the central nervous system. CNS Neurosci. Ther. 2013, 19, 76–82.
- Hu, G.; Gong, X.; Wang, L.; Liu, M.; Liu, Y.; Fu, X.; Wang, W.; Zhang, T.; Wang, X. Triptolide promotes the clearance of alpha-synuclein by enhancing autophagy in neuronal cells. Mol. Neurobiol. 2017, 54, 2361–2372.
- Eskandari, H.; Ghanadian, M.; Noleto-Dias, C.; Lomax, C.; Tawfike, A.; Christiansen, G.; Sutherland, D.S.; Ward, J.L.; Mohammad-Beigi, H.; Otzen, D.E. Inhibitors of alpha-synuclein fibrillation and oligomer toxicity in rosa damascena: The all-pervading powers of flavonoids and phenolic glycosides. ACS Chem. Neurosci. 2020, 11, 3161–3173.
- Gallardo-Fernandez, M.; Hornedo-Ortega, R.; Cerezo, A.B.; Troncoso, A.M.; Garcia-Parrilla, M.C. Melatonin, protocatechuic acid and hydroxytyrosol effects on vitagenes system against alpha-synuclein toxicity. Food. Chem. Toxicol. 2019, 134, 110817.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
558
Revisions:
2 times
(View History)
Update Date:
21 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No