Rheumatoid arthritis (RA), a chronic autoimmune disease, is characterized by irreversible cartilage and bone damage as well as processive disability, systemic complication, early death and socioeconomic costs. The basic pathological changes are chronic synovitis, pannus formation, and the progressive development of articular cartilage and bone damage. The incidence of RA is 0.5–1% and decreases significantly from north to south and from urban to rural areas in the northern hemisphere
[1]. Women are affected two to three times more often than men and incidence of new RA diagnoses peaks in the sixth decade of life. It is two to three times more prevalent in those with a family history of RA than those without, and the prevalence in twins is highly consistent, suggesting a genetic component to the cause of RA. In clinical practice, the manifestation of RA indicates as a symmetrical disease, initially affecting joints from small to larger
[2]. It not only primarily involves the joints, such as synovial inflammation and hyperplasia, but also includes extra-articular symptoms, such as cardiovascular, pulmonary, skeletal and psychological disorders
[3]. Recent papers have reported that meningitis, brain vasculitis and autoimmune encephalitis had been observed in RA patients
[4], which can cause severe cognitive decline, such as in short term memory, immediate and delayed episodic recall and phonemic fluency
[3]. RA is a chronic disease that causes musculoskeletal deformities, decreased physical function and cumulative complications that can lead to a subsequent decline in quality of life, which can be a sinking blow to individuals and families.
2. NO/NOS Pathway Overview
Nitric oxide (NO), a small gas molecule, is considered to play significant roles in the cardiovascular, immune and nervous systems and can be generated in the cytosol, the mitochondrial membrane and at the immunological synapse of T cells. NO can diffuse freely in membranes so that it can act as message molecule to mediate various biological functions, such as regulating systolic and diastolic functions of blood vessels, T cells signaling pathways, inflammation, oxidative stress, mitochondrial functions and apoptosis. It mediates signal transduction by regulating Ca2+ signaling pathways, by changing the formation of immunological synapses, or through the modification of intra-cellular proteins.
nitric oxide synthases (NOS) is associated with generation of NO, which can transform L-arginine to L-citrulline and then produce NO. NOS include three isoforms encoded by different genes, neuronal nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS)
[5]. Molecular oxygen, reduced nicotinamide adenine dinucleotide phosphate (NADPH) and L-arginine serve as the substrate and co-substrates, respectively, for all isoforms of NOS
[5]. All isoenzymes require the cofactors flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN) and 6R-l-erythro-5,6,7,8-tetrahydrobiopterin (BH
4). The reductase domain of this monomer consists of NADPH, FMN and FAD binding sites, whereas the oxygenase domain consists of BH
4, O
2 and L-arginine binding sites and a heme group for NOS dimerization (
Figure 1). There is also a calmodulin binding domain located between the reductase and oxygenase domains
[6].
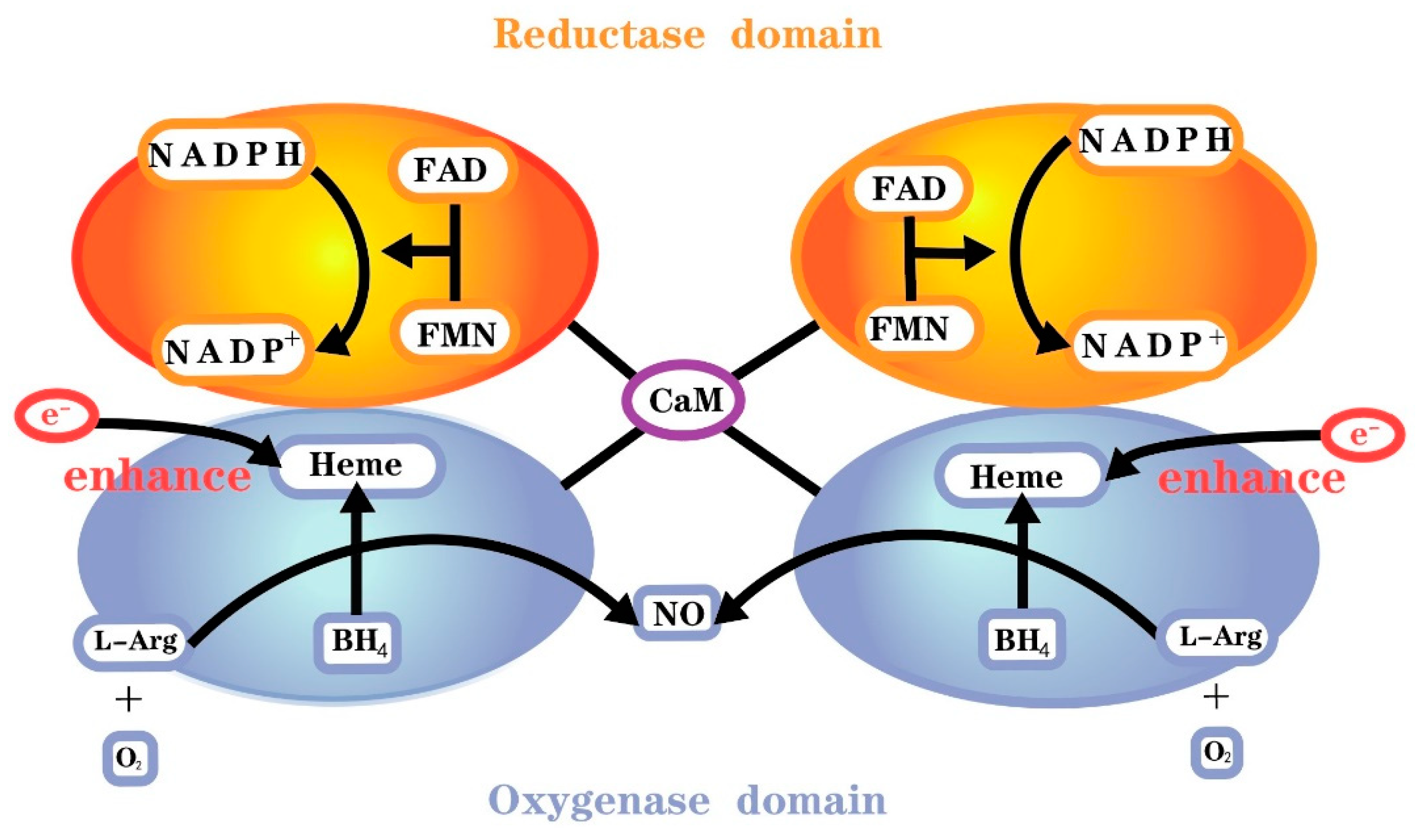
Figure 1. Functional nitric oxide synthase structure.
The first step of NO synthesis is the transfer of electrons from reduced NADPH to FAD and FMN by NOS monomers and the reduction of molecular oxygen to superoxide (O
2−)
[7]. However, the reductase domain separated by the two monomers needs to be bound to calmodulin (CaM) to enhance electron transport in the reductase domain. All isoforms of NOS bind calmodulin, hence presenting catalytic activity, but the calmodulin binding domains of NOS differ among isoforms. Calmodulin binding in nNOS and eNOS requires elevated Ca
2+, and calmodulin binds iNOS with high affinity even in the absence of Ca
2+ [8]. The second step is binding to the cofactor BH
4 or the substrate L-arginine. The two NOS monomers need to form a complete functional dimer in the presence of haem to catalyze NO formation.
nNOS is expressed in the brain, spinal cord, sympathetic ganglia, skeletal muscle, epithelial cells and vascular smooth muscle. The enzyme activity is modulated by calcium and calmodulin. Different forms of nNOS exists in in different cells with distinct functions. In the central nervous system, nNOS is involved in regulating physiological functions such as memory, learning, neurogenesis and mediating long-term regulation of synaptic transmission. In addition, NO formed by nNOS in the central nervous system is involved in the central regulation of blood pressure
[9]. In the periphery, many smooth muscle tissues are innervated by nitro-energic nerves, which are nerves that contain nNOS and produce and release NO.
eNOS is mostly expressed in endothelial cells, while it is also found in certain brain neurons, platelets, cardiomyocytes and renal tubular epithelial cells. Similar to nNOS, Ca
2+ activated calmodulin is crucial for controlling the activity of eNOS
[10]. Additionally, a number of other proteins engage in interactions with eNOS and control its activity. For instance, heat shock protein 90 (HSP90) functions as an allosteric regulator that encourages eNOS coupling, as well as as an eNOS-related activator. The Caveolae coat protein Caveolin-1 is a tonic inhibitor of eNOS activity, which can interact with the eNOS fraction. Recruitment of calmodulin and HSP90 to eNOS dislocates caveolin-1 from the enzyme, resulting in enzyme activation
[11]. However, stimulation that does not result in a long-lasting rise in intracellular Ca
2+ can trigger another independent method of eNOS activation.
iNOS is not normally expressed in cells, but its expression can be induced by bacterial lipopolysaccharides, cytokines and other factors, and it can be stimulated in almost any cell or tissue. Once expressed, iNOS is continuously activated and not regulated by intracellular Ca
2+ concentration. When induced in macrophages, inducible NOS produces large amounts of NO, which results in cytotoxicity
[12]. Increased NO can inhibit important enzymes that have iron in their catalytic centers because of its affinity for protein-bound iron, including ribonucleotide reductase, which is involved in DNA replication;
cis-aconitase; and iron-sulfur cluster dependent enzymes involved in mitochondrial electron transport
[13].
3. The Pathological Changes of RA
Histological Changes
Synovitis is the key to the pathophysiology of RA, and synovial structure changes as the RA progresses. In the early stage of onset, tissue edema and fibrin deposition are evident, with clinical manifestations of joint swelling and pain
[14]. The characteristic of this stage is the entry of leukocytes and neutrophils from the blood into the synovial tissues (
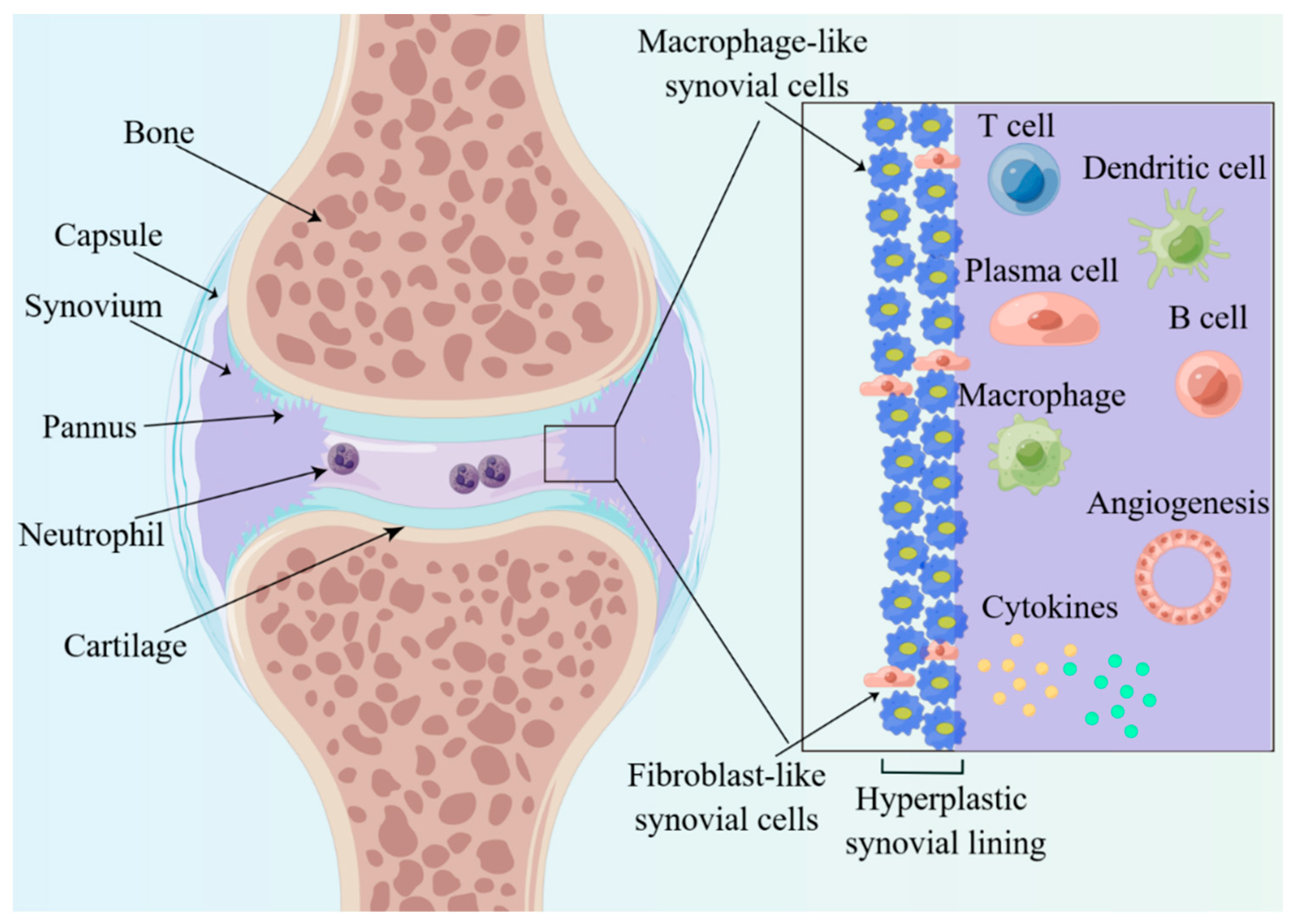
Figure 2). In the short term, the synovial membrane becomes hypertrophic, usually forming 10 or more cells consisting of macrophage-like (type A) and fibroblast-like (type B) synovial cells (
Figure 2), and forms many villous projections that protrude into the joint cavity or invade the cartilage and subchondral bone. The villous projections, also called pannus, damage and produce erosion of the articular cartilage, which is the distinctive pathological basis of destruction, deformity and dysfunctions of joints. Significant changes also take place in terms of the number and the sub-cellularity of immune cells, with a marked infiltration of single nucleated cells, such as plasma cells, macrophage, B cells and T cells. Moreover, there exist changes in the expression of cell surface adhesion molecules, proteases, protease inhibitors and many cytokines in synovitis.
Figure 2. Histological features of inflamed joints of RA. The enlarged synovial lining layer consists of macrophage-like and fibroblast-like synovial cells and then forms many villous projections called pannus. Significant changes also take place, with a marked infiltration of cytokines, plasma cells, macrophages, neutrophils, B cells and T cells.
4. The Pathogenesis of RA
4.1. RA and Autoimmunity
The tissue destruction caused by the autoimmune effects of RA manifests as synovitis, which is jointly initiated and maintained by the combination of the activation of various immune cells, including the different dendritic cells (DCs) subtypes, T-cells, macrophages, B-cells, neutrophils and other cells such as fibroblast and osteoclasts
[15].
Accumulating evidence indicates that the concentration of DCs in the plasma of RA patients results from the recruitment of DCs in the flamed joints. This distribution and changed function of DCs contributes to the development of RA. Activated DCs are capable of producing IFN-α, IFN-β, IL-18 and IL-23, increasing the generation of autoantibodies by expression of anti-apoptotic B cell activating factor, and promoting the presentation of autoantigens to T cells, which potentiates the autoimmune response and maintains the inflammation. Activated T cells can secrete IL-2, IFN-γ and TNF-β and Th1 cells can activate other immune cells, such as macrophages and B cells. Th 17 cells, activated in synovial joints, are capable of recruiting neutrophils, activating immune cells and enhancing osteoclastogenesis
[16]. Macrophages are activated and then generate pro-inflammatory cytokines including IL-1β, IL-6 and TNF-α, which can accumulate and activate other immune cells in the synovium
[15]. Activated fibroblasts, together with other accumulated activated immune cells, promote osteoclastogenesis through the nuclear factor κB ligand receptor. In healthy people, there exist a balance between bone resorption by osteoclasts and bone formation by osteoblasts, while the balance is disrupted in RA patients, whose bone resorption becomes excessive. Increased expression of iNOS genes and proteins is seen under hypoxic conditions or oxidative stress, which promotes osteoclast differentiation
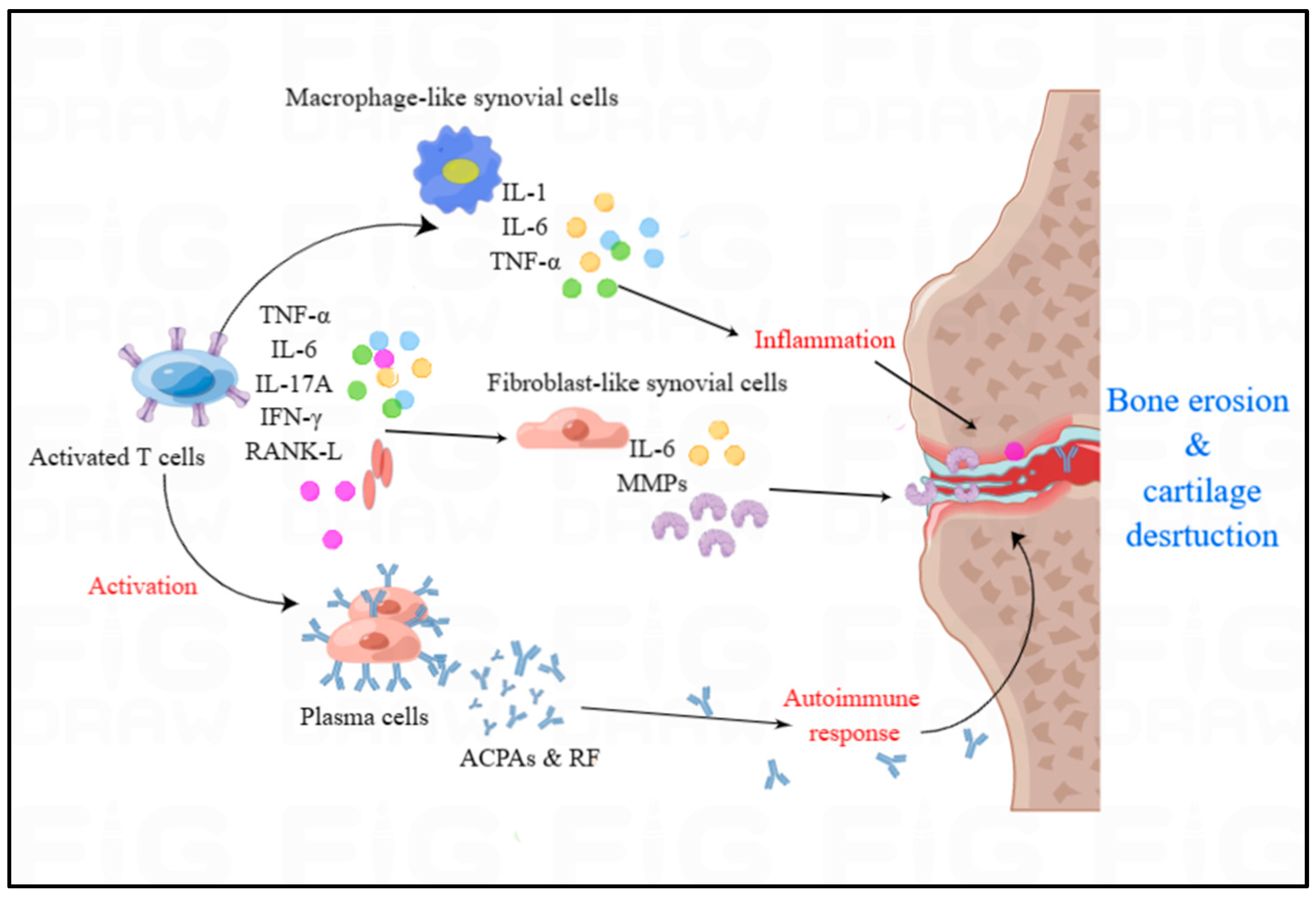
[17]. The high concentration of RA specific antibodies, such as rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPAs), is associated with joint damage and high mortality (
Figure 3), which there is no effective treatment to eliminate at present
[15].
Figure 3. The overview of immune cells and cytokines in RA. Autoimmunity and inflammatory cytokines play significant role in the development of RA. Activated T cells predominantly generate TNF-α, IL-6, IL-17A, IFN-γ, and RANK-L, which can activate the plasma cells, fibroblast-like synovial cells and macrophage-like cells. Macrophage-like cells can produce IL-1, IL-6 and TNF-α to induce the inflammation in the synovium of RA. Activated plasma cells can release the autoantibodies including ACPAs and RF to trigger autoimmune responses in RA.
4.2. RA and Cytokines
Chronic inflammation and synovitis with joint swelling are the typical characteristics of RA
[18]. Some studies have documented evidence for elevated cytokines concentrations in the affected tissues of pre-RA and RA patients compared with healthy subjects
[19]. In RA, the concentration of pro-inflammatory cytokines is higher than anti-inflammatory cytokines, which can result in RA synovia
[19]. Macrophage-like synoviocytes and fibroblast-like synoviocytes are the main sources of cytokines. Macrophage-like synoviocytes may result in the overproduction of proinflammatory cytokines including IL-1, IL-6 and TNF-α, thus promoting persistent inflammation and joint destruction (
Figure 3). Fibroblast-like synoviocytes express IL-6, MMPs, prostaglandins and leukotrienes, which manifests an aggressive inflammatory, stromal regulatory and invasive phenotype in the synovial compartment. The inflammatory environment in the synovial compartment is induced and maintained by the cytokine and chemokine network, including tumor necrosis factor TNF-α, IL-6, IL-17A, interferon IFN-γ and receptor activator of nuclear factor κB ligand (RANK-L) (
Figure 3), which can activate endothelial cells and accumulate immune cells in the synovial compartment to trigger and regulate inflammation via the binding of cytokines to cell receptors. Recently, some successful cellular RA models induced by TNF-α have induced fibroblast-like synoviocytes
[20].
5. NOS/NO Pathway in RA Pathogenesis
NOS and NO are critical signal molecules with various physiological and pathological effects, which regulate a variety of cellular events. NO, produced by NOS, is an endogenous vasodilator and intercellular messenger in the cerebral and peripheral blood flow
[21]. NO plays an important role in self-protective inflammatory and immune responses, but excessive endogenous NO may cause inflammatory diseases such as RA. There are several kinds of cells can generate NO, such as macrophages, fibroblasts, neutrophils, endothelial cells, osteoblasts and osteoclasts in the synovium
[22]. It is reported that indications of NO-dependent tissue injury and overproduction of endogenous NO synthesis are present in RA, which suggests that overproduction of NO is important in RA
[23].
5.1. NOS/NO Pathway Involved in RA Autoimmunity
NO plays an important role in the immune system, and can be produced by many different immune cell types, including dendritic cells, NK cells, macrophages, mast cells, eosinophils and neutrophils. NO and these immune cells are involved in the development of RA.
T cell functions are considered to have a key role in the pathogenesis of RA. NO has been proven to regulate T cell functions in normal ranges, but excessive NO can result in T cell dysfunction. In mammals, NO regulates T cell activation, mitochondrial membrane potential and mitochondrial biogenesis. Based on the current reports, T helper 1 (Th 1)/Th 2 and Th 17/Treg imbalance can potentiate the activity of RA. Many studies show that eNOS-derived NO is able to enhance the signal transduction of antigens to T cell receptor at the immunological synapse, promoting the activation of T cells
[24]. After activation, CD4 T cells proliferate and differentiate into Th 1 and Th 2 cells. Th 2 cells express various anti-inflammatory cytokines with protective effects for humans. NO can selectively promote the proliferation of Th 1 and influence the Th 1/Th 2 balance, which is essential in chronic inflammatory disease.
5.2. NOS/NO Pathway Involved in RA Oxidative Stress
More and more evidences suggest that oxidative stress can accelerate the development of RA. Oxidative stress markers, NO and peroxides (PO) also constitute the major pathophysiological factors of RA. NO can react with superoxide (O
2−) to produce peroxynitrite (ONOO
−), affecting the cytoplasmic redox balance. The excessive accumulation of free radicals could trigger oxidative stress, which contributes to the pathogenesis of RA
[25]. Reactive oxygen synthase (ROS) and reactive nitrogen synthase (RNS) can participate in a variety of cellular activities.
The excessive accumulation of harmful free radicals and superoxide can trigger an imbalance of oxidants and antioxidants, resulting in oxidative stress. ROS generate oxygen in an oxidative stress environment. The reaction rate of O
2− with NO is 3 to 4 times faster than that of superoxide dismutase. Therefore, if the O
2− concentration increases or NO levels rise excessively, they will react to produce the massively cytotoxic substance ONOO
− [26]. ONOO
− can cause joint cell degradation, including lipid peroxidation, protein dysfunction, and DNA damage, thus exacerbating the condition of RA
[27]. Oxidative stress can decrease NO levels and NO bioavailability by producing ONOO
−, thus further exacerbating the oxidative stress condition and finally forming a vicious circle. One recent paper demonstrated that iNOS promote the release of ONOO
−.
5.3. NOS/NO Pathway Involved in RA Inflammatory Cascade
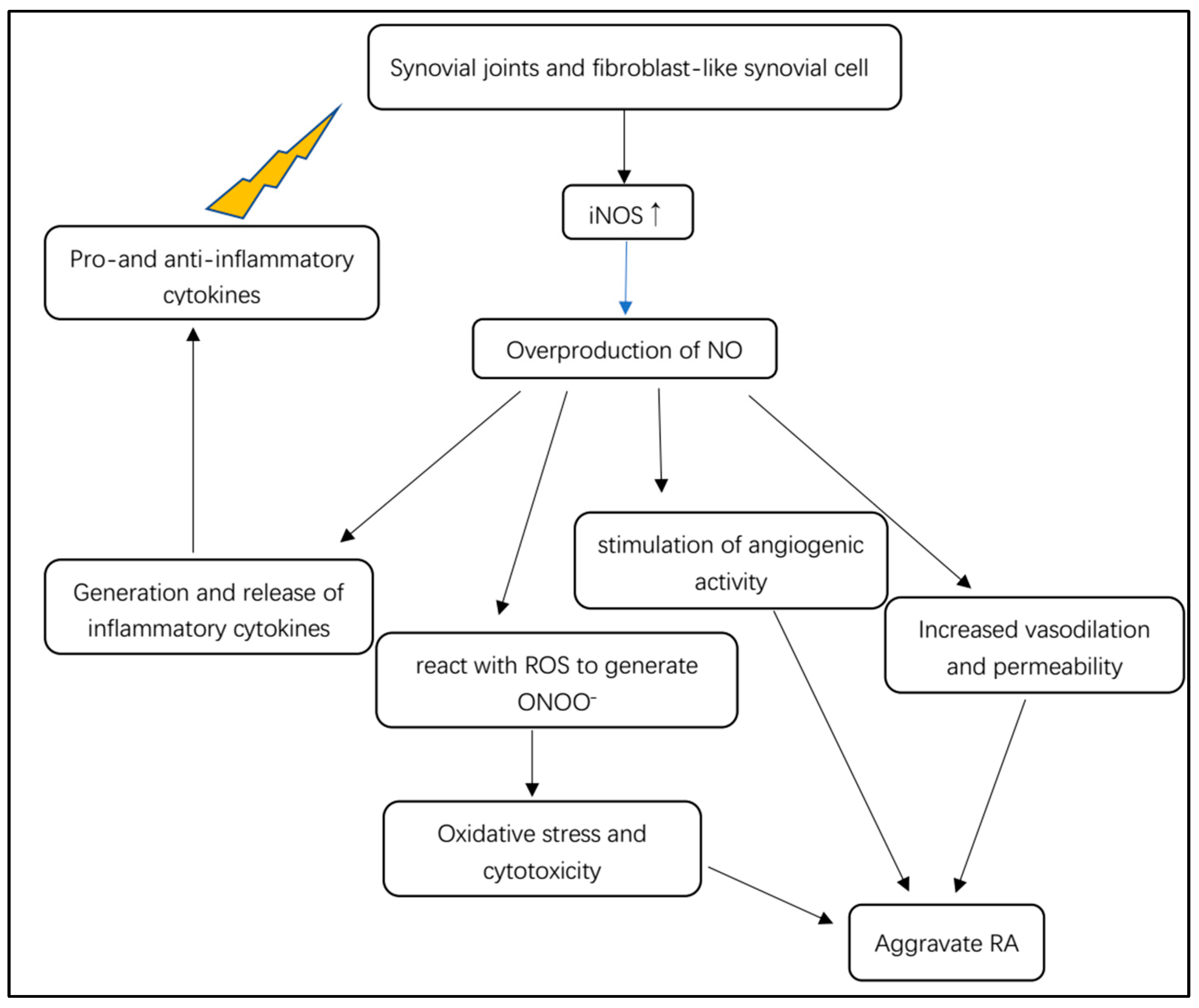
In rheumatoid arthritis, NO is involved in inflammation, angiogenesis and tissue degradation (
Figure 4). Pro- and anti-inflammatory cytokines stimulate synovial macrophages and fibroblast-like synovial cells to express iNOS (
Figure 4)
[19]. iNOS stimulated by cytokines is responsible for the local overproduction of NO in joints affected by RA
[19]. iNOS is considered to be mainly expressed in unhealthy states, such as inflammatory microenvironments, where it can be activated or triggered by various cytokines such as TNF-α, TNF-β and IL-6. Furthermore, cell senescence can express a special pro-inflammatory phenotype, indicating that senescent synoviocytes can secrete inflammatory factors such as TNF-α, TNF-β and COX-2
[28]. After immunological or microbial stimulation, iNOS can be induced to remain active to produce excessive NO, mediating the generation and release of inflammatory cytokines
[12].
Figure 4. Overview of the pathological mechanism of iNOS-dependent NO for RA.
5.4. NOS/NO Pathway Involved in RA Circadian Rhythm
The progression of RA is closely linked to the chronobiology changes in the production of certain hormones and inflammatory mediators that affect the course of the disease and the outcome of treatment. RA is perceived as a disease disrupting the circadian rhythm of steroid hormone or cytokine production. In recent years, circadian rhythm disturbance of the inflammatory process in RA has been recognized as one of the important inflammatory mechanisms that triggers inflammation, destruction and proliferation of synovial joints
[29]. eNOS and soluble Toll-like receptor 2 (sTLR2) are involved in the regulation of angiogenesis, osteoclast-genesis and immune response
[30]. The main pathogenesis of RA is controlled by the biological clock gene angiogenesis
[14].
5.5. NOS/NO Pathway Involved in RA Complications
5.5.1. The Effect of NOS/NO Pathway in Cardiovascular Manifestation of RA
Cardiovascular disease (CVD) is regarded as a common complication in RA patients. RA increases the risk of CVD, which accounts for the highest morbidity and mortality observed in RA patients. Endothelial dysfunction resulting from impaired nitric oxide synthesis increases the risk of the cardiovascular disease. eNOS uncoupling increases cardiovascular mortality associated with autoimmune rheumatic diseases. Oxidative stress has a significant role in endothelial dysfunction, which is also identified to be caused by eNOS uncoupling (
Figure 5). The conversion of uncoupled eNOS into superoxide-producing enzyme can decrease the production level of NO and enhance pre-existing oxidative stress, which plays a critical role in the process of atherosclerosis
[31].
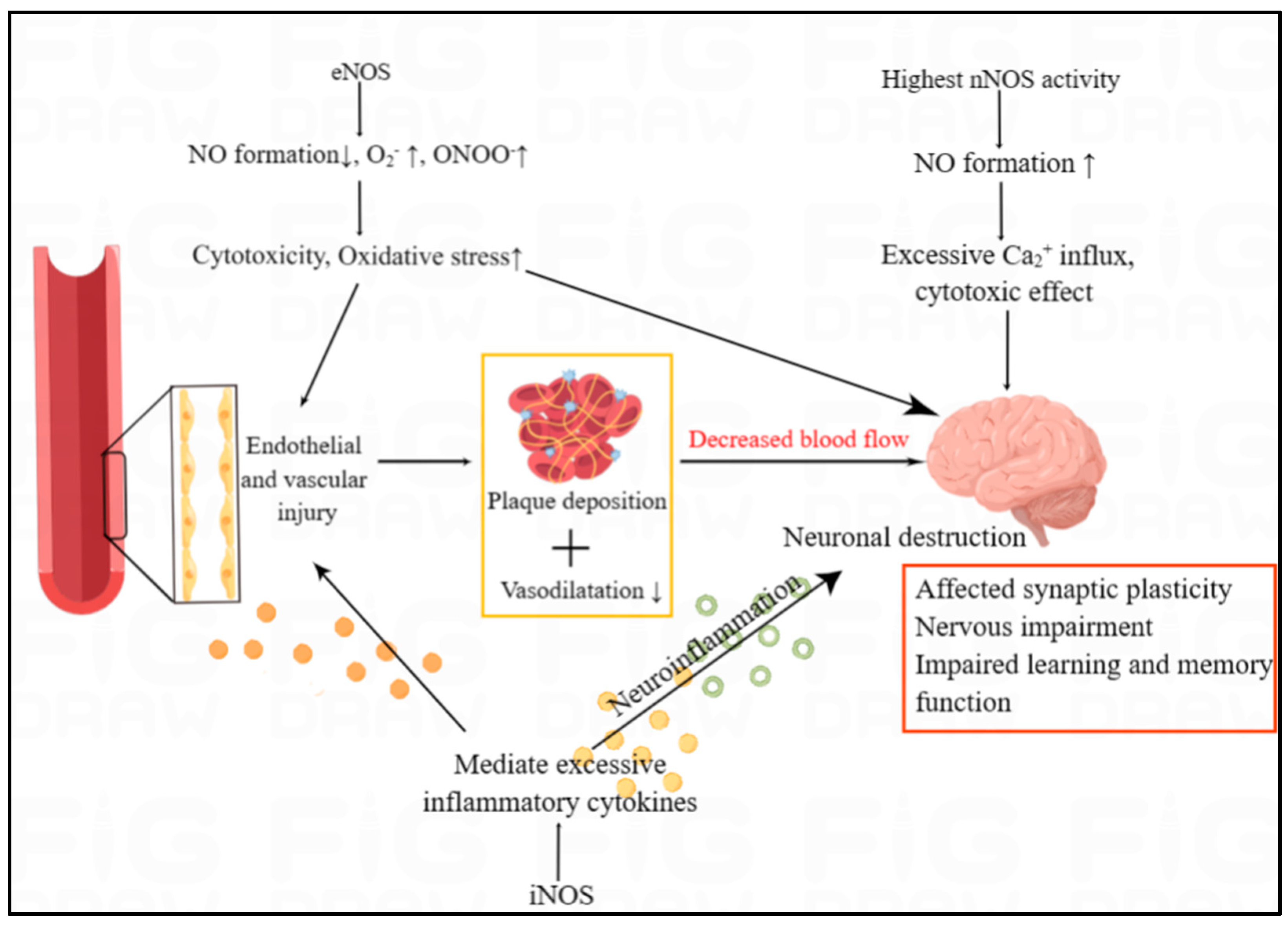
Figure 5. The overview of NOS/NO in the cardiovascular and neurological manifestation of RA. The three NOS isoforms, with entirely different functions, jointly influence brain function. In humans, nNOS activity is highest in the brain, affecting the synaptic plasticity. In both the endothelium and the brain, iNOS and eNOS can trigger inflammatory responses and high oxidative stress, which can lead to endothelial injury and neuronal destruction. Endothelial and vascular injury are associated with plaque deposition and decreased vasodilation, which results in the decreased blood flow to brain.
5.5.2. The Effect of NOS/NO Pathway in Neurological Manifestations of RA
Brain lesion is rarely reported in RA, but cognitive decline was studied in the article of Pankowski et al.
[32]. Neurological manifestations of RA range from mild hand sensory abnormalities and carpal tunnel syndrome to cognitive dysfunction
[33][34]. Compressive neuropathy, particularly carpal tunnel syndrome, is the most common abnormality of the peripheral nervous system in RA
[33]. The three NOS isoforms, with entirely different functions, jointly influence brain function. It has been reported that the reduction of eNOS activity as well as the overactivation of nNOS and iNOS can lead to brain lesion. Several studies have shown that NO has both neuroprotective and neurotoxic effects, which depends on its bioavailability, concentration and the redox state of the microenvironment of the body
[35]. nNOS is abundant in central and peripheral neurons, and nNOS-derived NO play an important role in learning and memory ability via participating in changing the concentration of Ca
2+. Ca
2+ has a significant role in affecting synaptic plasticity
[36]. iNOS can mediate excessive release of inflammatory cytokines which can leads to neuroinflammation, thus continuously resulting in neuronal destruction (
Figure 5).
6. Disease-Modifying Antirheumatic Drugs Targeting NOS/NO Pathway
6.1. Conventional DMARDs
In recent years, with the in-depth research on some conventional drugs for the treatment of RA, the specific mechanisms of action and therapeutic effects have been explored. The elucidation of the mechanism of DMARDs will help to control adverse reactions. For example, Etanercept can significantly alleviate arthritis, which may be achieved by increasing eNOS phosphorylation and expression, decreasing arginase-2 and subunit of NOX p22-phox expression and decreasing O
2− production. In addition, Etanercept has been found to exert pleiotropic effects on endothelial pathways, improving endothelial dysfunction and reducing risk factors of cardiovascular disease, with significant increases in systolic blood pressure and heart rate
[37]. Indometacin (IDMT) is an analgesic used to relieve pain in RA. IDMT was found to be able to alleviate RA-induced endothelial cell damage through two different pathways, higher concentrations of IDMT through the IKK-COX-2/TNF-α pathway, and lower concentrations through the PPARγ-Akt-eNOS pathway. Furthermore, the mechanism of another conventional antiarthritic drug, Auranofin (AF), has been elucidated. AF is an anti-inflammatory gold compound that has been used to treat RA for more than four decades. AF can inhibit the upregulation of NOX-4 and the translocation of NF-κB in RA macrophages by binding to Toll-like receptors. AF can suppress expression levels of inflammatory stimulants iNOS and reduce NO levels through the induction of pro-inflammatory proteins and mRNA by the upstream regulatory factor NF-κB
[38].
6.2. Novel DMARDs
New synthetic anti-RA drugs mostly achieve anti-inflammatory effects by targeting specific receptors. An active proinflammatory mediator with several functions, macrophage migration inhibitory factor (MIF), contributes to the development of RA. MIF inhibitor compound 3-[(biphenyl-4-ylcarbonyl) carbamothioyl] amino benzoic acid (Z-590) has potent anti-arthritis effects achieved by inhibiting macrophage activation and significantly improving joint inflammation and articular cartilage damage. Z-590 can significantly inhibit the production of NO through suppressing expression of iNOS in RA macrophages in and in vitro model
[39]. By activating NO/cGMP/ATP-sensitive potassium channels without impairing inflammatory processes, the 7-nicotinic acetylcholine receptor (7nachRs) agonist choline can control inflammatory hyperalgesia, suggesting its potential therapeutic potential for the treatment of RA pain
[40]. Cannabinoid receptor 2 (CB2) has powerful anti-inflammatory effects, and a selective agonist of CB2, 4-quinolone-3-carboxamides (4Q3C), can significantly improve arthritis and reduce bone erosion. Treatment with 4Q3C reduced the expression of iNOS
[41]. Soluble epoxide hydrolase (sEH) can rapidly convert endogenous anti-inflammatory compounds, for example, epoxyeicosatrienoic acids, into less active forms. Thus, inhibition of sEH enzymes is a potent strategy to reduce nociception and inflammation. SEH enzyme inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) can attenuate arthritic hyperalgesia and inflammation via peripheral intervention. TPPU significantly reduced iNOS protein expression and local concentrations of proinflammatory cytokines
[42]. 2-Deoxyglucose (2-DG), a glycolysis inhibitor, was shown to have anti-arthritic properties and can reduce cell infiltration, synovial hyperplasia and bone erosion in RA patients. Its therapeutic mechanism has recently been found to be related to repression of NF-κB in peritoneal macrophages in rat model of RA, thereby promoting the transformation of macrophages type from M1 to M2. By regulating macrophage polarization as well as metabolic changes, 2-DG increases arginase 1 (Arg1) levels and decreases iNOS expression
[43].
 +1 credit
+1 credit