Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Samuel Lee | -- | 2286 | 2023-07-19 02:10:21 | | | |
| 2 | Fanny Huang | Meta information modification | 2286 | 2023-07-19 07:34:46 | | | | |
| 3 | Sangyoun Hwang | Meta information modification | 2286 | 2023-07-20 01:27:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hwang, S.Y.; Liu, H.; Lee, S.S. Dysregulated Calcium Handling in Cirrhotic Cardiomyopathy. Encyclopedia. Available online: https://encyclopedia.pub/entry/46947 (accessed on 25 July 2026).
Hwang SY, Liu H, Lee SS. Dysregulated Calcium Handling in Cirrhotic Cardiomyopathy. Encyclopedia. Available at: https://encyclopedia.pub/entry/46947. Accessed July 25, 2026.
Hwang, Sang Youn, Hongqun Liu, Samuel S. Lee. "Dysregulated Calcium Handling in Cirrhotic Cardiomyopathy" Encyclopedia, https://encyclopedia.pub/entry/46947 (accessed July 25, 2026).
Hwang, S.Y., Liu, H., & Lee, S.S. (2023, July 19). Dysregulated Calcium Handling in Cirrhotic Cardiomyopathy. In Encyclopedia. https://encyclopedia.pub/entry/46947
Hwang, Sang Youn, et al. "Dysregulated Calcium Handling in Cirrhotic Cardiomyopathy." Encyclopedia. Web. 19 July, 2023.
Copy Citation
Cirrhotic cardiomyopathy is a syndrome of blunted cardiac systolic and diastolic function in patients with cirrhosis. Since contractility and relaxation depend on cardiomyocyte calcium transients, any factors that impact cardiac contractile and relaxation functions act eventually through calcium transients. In addition, calcium transients play an important role in cardiac arrhythmias.
cirrhotic cardiomyopathy
L-type calcium channel
calcium transient
1. Introduction
Cardiovascular abnormalities in cirrhosis include hyperdynamic circulation and cirrhotic cardiomyopathy (CCM); the former is the combination of decreased peripheral vascular resistance and increased cardiac output whereas the latter is a syndrome of cardiac contractile dysfunction in the absence of a primary cardiac condition [1][2][3]. There are two sets of diagnostic criteria for CCM: the World Congress of Gastroenterology (WCG, 2005) criteria and the Cirrhotic Cardiomyopathy Consortium (CCC, 2019) criteria. The latter is based on advanced imaging methods. Whether the new criteria are superior to the old is not yet settled, although the current consensus seems to favor the new criteria.
The pathophysiology of the cardiovascular system is complex. In cirrhotic patients and animal models, the abnormal architecture of the liver causes portal venous hypertension, and the congested gut results in increased intestinal permeability. These pathophysiological changes trigger bacterial overgrowth, translocation, and increased bacteria-derived products in their circulation, including endotoxins that elevate systemic pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin-1β (IL-1β) and interleukin-6 (IL-6). Thus, patients with cirrhosis and portal hypertension manifest an inflammatory phenotype that leads to increased vasodilators such as nitric oxide, carbon monoxide, prostaglandins, bile acids, and glucagon. Therefore, the peripheral vasculature is dilated.
It is well noted that the sympathetic nervous system (SNS) tone is increased in cirrhotic patients (serum concentration of norepinephrine is increased) [4]. The increased activity of the SNS and elevated serum concentrations of vasoconstrictive catecholamines are a compensatory reaction to vasodilatation, not its cause. In cirrhosis, vasodilators predominate over vasoconstrictors, and, therefore, the phenotype of cirrhotic patients and animal models is systemic vasodilatation. Because of the baseline peripheral vasodilatation of cirrhotic patients, cardiac dysfunction is subclinical at rest, which results in delayed or missed diagnosis in clinically stable patients with cirrhosis. However, when patients face cardiovascular challenges such as liver transplantation, transjugular intrahepatic portosystemic shunt (TIPS) insertion, hemorrhage, and vasoactive drugs [1], cardiac dysfunction manifests overtly.
There are numerous pathogenic mechanisms underlying CCM which broadly comprise two distinct pathways: factors related to (a) liver dysfunction with synthetic failure/dysregulation of substances such as proteins, lipids, lectins, hormones, etc., and (b) portal hypertension with gut congestion and induction of the inflammatory phenotype.
Uhlig and coworkers [5] demonstrated that serum TNFα and cardiac inflammatory cells are significantly increased in cirrhotic rats. Researchers' lab [6] showed that Fas protein expression and PARP cleavage, which are indices of apoptosis, are significantly increased in the cirrhotic hearts of rats. Moreover, anti-FasL monoclonal antibody injection in BDL-mice improved systolic and diastolic dysfunction. Thus, the study implies that apoptosis plays an important pathogenic role in murine cardiac dysfunction. Another study [7] on oxidative stress revealed that oxidative stress-related parameters are increased and antioxidant regulation is decreased in cirrhotic hearts. Erythropoietin, an antioxidant, significantly downregulated oxidative stress and reversed impaired cardiac function. Structural changes in cirrhotic rat hearts included alterations in myosin heavy chain (MHC) isoforms, leading to a dominance of weaker-contracting β-MHC over the more powerful α-MHC [8] and increased cardiac fibrosis [5].
Among the ions involved in cardiac dysfunction in cirrhosis, calcium transients play an essential role.
2. Calcium Transport in Cardiac Excitation–Contraction Coupling
The cardiac action potential is a brief change in voltage across the plasma membrane of cardiac myocytes. In a normal heart, the action potential of the ventricular myocardium triggers cardiac contraction. There are five phases in the action potential of the ventricular myocardium. Researchers' previous studies have demonstrated abnormalities of two ion transients, calcium [8][9] and potassium [10], in rat cirrhotic ventricular myocytes.
Calcium entering the cytoplasm via L-type calcium channels triggers the opening of calcium release channels (ryanodine receptor), leading to calcium release from the sarcoplasmic reticulum (SR). The released calcium binds troponin C, which enables the movement of tropomyosin and thus exposes the myosin binding site on the thin filaments (Figure 1) [11]. A cross-bridge is then formed between the thick and thin filaments which results in cardiac contraction [12]. After contraction, both voltage-gated calcium channels and calcium release channels are closed, and the calcium is removed from the cytosol via the sarcoplasmic-endoplasmic reticulum calcium-ATPase (SERCA) to the SR and via the sodium–calcium exchanger (NCX) outside of the cardiomyocyte [13]. In the steady state, the amount of calcium entering the cell must be equal to that extruded in each cardiac cycle. If not, the cell would either gain or lose calcium [14]. Calcium transport is abnormal in cirrhotic cardiomyopathy [8][9].
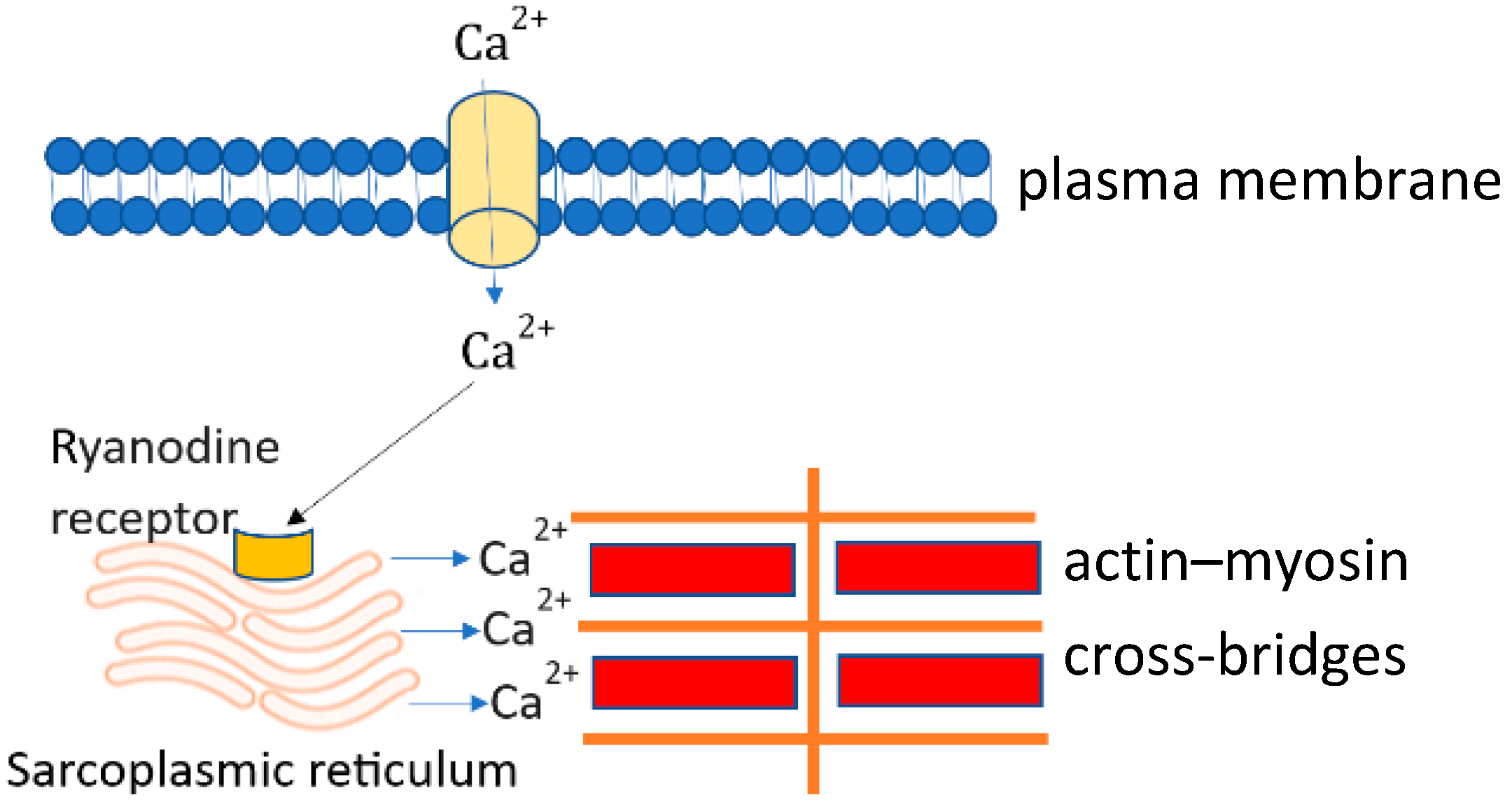
Figure 1. Calcium transients in cardiomyocytes.
At phase 2 of the action potential, calcium enters the cytosol through L-type calcium channels which activate ryanodine receptors (calcium-induced calcium release channels). Activation of ryanodine receptors triggers the release of calcium from the sarcoplasmic reticulum. Calcium released mainly from the sarcoplasmic reticulum combines with the troponin complex and triggers actin–myosin cross-bridge linking, and thus cell contraction. This process is called excitation–contraction coupling.
3. Abnormal Membrane L-Type Calcium Channels in Cirrhotic Cardiomyopathy
Calcium is an essential second messenger in all living organisms. From Caenorhabditis elegans to mammals, calcium is mandatory for body functions, locomotion, and neural activities [15]. In the late nineteenth century, Sidney Ringer published his observation which demonstrated that calcium is essential for cardiac contraction: the removal of calcium from the perfusion buffer of the frog heart halted cardiac contraction [16]. The force of cardiac contractility is calcium concentration-dependent [8]. This experiment implied that external calcium is required for cardiac systole. L-type calcium channels are the channels through which calcium outside the cell enters the cytosol. L-type calcium channels are essential for the initiation and regulation of excitation–contraction (EC) coupling in cardiomyocytes. The rapid entry of calcium via these channels triggers the release of intracellular calcium from ryanodine receptors embedded on the surface of the SR. The released calcium activates the myofilaments and finally triggers the contraction of cardiomyocytes [11]. Any abnormality of L-type calcium channels impacts the amount of cytosolic calcium prior to the contraction and therefore affects contractility.
The abundance and current of L-type calcium channels are decreased following the development of cardiac dysfunction. Mukherjee et al. [17] studied pacing-induced chronic heart failure in pigs. They reported that two weeks after pacing (240 bpm), the content of L-type calcium channels was significantly decreased compared to controls (Bmax. 149 ± 16 vs. 180 ± 12 fmol/mg, p < 0.03) as was the current of L-type calcium channels (2.47 ± 0.10 vs. 3.63 ± 0.25 pA/pF, p < 0.02). Researchers' study [9] on cirrhotic cardiomyopathy in rats showed a similar pattern to the Mukherjee study. In Sprague Dawley rats, a model of cirrhotic cardiomyopathy was created by bile duct ligation (BDL). Researchers demonstrated that the protein expression of L-type calcium channels in cirrhotic hearts is significantly reduced compared with controls, and the peak inward calcium current was significantly reduced (−4.9 ± 0.3 pA/pF vs. −7.3 ± 0.9 pA/pF, p < 0.0001) [9]. At all membrane potentials examined, the current densities of calcium influx entering via L-type calcium channels were consistently lower in cardiomyocytes measured from cirrhotic animals than that from sham controls.
The response of peak inward calcium current to maximal isoproterenol stimulation was also significantly lower. Protein expression and messenger RNA transcription for RyR2, SERCA2, and calsequestrin were quantitatively unchanged [9].
4. Abnormal Intracellular Calcium Handling System
It is now clear that the density and function of L-type calcium channels are reduced in cardiomyocytes from cirrhotic rats. This abnormality impacts the calcium transients in the intracellular calcium handling system, mainly ryanodine receptors (RyRs). RyR is a homotetrameric intracellular calcium release channel that is composed of four 565-kDa protomers and is located on the SR in cardiomyocytes and other cell types [15]. The name derives from its ability to bind ryanodine, a component of the Ryania speciosa plant used by South American Indigenous people in blow darts to paralyze prey. When ryanodine binds RyR channels, they induce an open state which results in an uncontrolled release of calcium from the SR, leading to muscle tetany. The flux of cytosolic calcium transients, calcium influx, and efflux should be balanced to maintain normal contractile function. RyR dysfunction is associated with a variety of conditions such as cardiac arrhythmias and heart failure. Although there are no structural changes in the intracellular calcium handling system in cirrhotic cardiomyopathy [9], the intracellular calcium handling system is disorganized by phenomena such as dispersion.
The functional unit (calcium release unit) is composed of the L-type calcium channel and ryanodine receptor clusters (RyR clusters) which play an essential role in regulating calcium transients. In an in vivo study, Kolstad and co-workers [18] showed that RyR clusters are abnormal in structure and function in cardiomyocytes from heart failure. They isolated and tested the RyR status in cardiomyocytes from rats with infarction-derived heart failure. Using super-resolution imaging, they observed that RyR clusters in failing cardiomyocytes showed dispersion, which resulted in more numerous, smaller clusters. Compared with sham controls, RyR clusters from heart failure produced weaker calcium sparks, calcium uptake was decreased, releasable SR calcium content was reduced, and leakage was increased. All these changes resulted in slow calcium kinetics leading to a weakened cardiac contraction in heart failure. This group further tested the role of the over-stimulation of beta-adrenergic receptors in the dispersion of RyRs [19]. They used isoproterenol to stimulate ventricular cardiomyocytes isolated from rats. They showed that long time stimulation with isoproterenol caused the dispersion of RyR clusters in the SR. The dispersion of RyR reduced calcium spark fidelity and magnitude. Furthermore, the dispersion of RyR also increased the ‘silent’ calcium leak. These changes resulted in smaller and desynchronized calcium transients, which are hallmarks of cardiac dysfunction.
It is well known that the sympathetic nervous system is over-activated in patients with cirrhosis. In the early 1960s, Shaldon et al. [20] measured the level of catecholamines, an index of sympathetic nervous activity, and showed that this is increased in portal venous plasma. Many other studies confirmed that the sympathetic nervous system is overactivated in patients with cirrhosis [21]. The overactivated sympathetic nervous system stimulates beta-adrenergic receptors which may cause dispersion of RyR, thus leading to contractile dysfunction in the cirrhotic heart.
The ventricular systolic force depends on the magnitude of the cytosolic calcium transient and the sensitivity of the contractile proteins to the calcium. Researchers showed that cellular calcium transients are reduced in cardiomyocytes from BDL-cirrhotic rats, which explains the decreased systolic force in these animals [8]. Another factor in the decreased sensitivity of myofilament to calcium in cirrhotic cardiomyocytes is the shift from an alpha-myosin heavy chain (α-MHC) to β-MHC. The α-MHC isoform contracts more vigorously than the β-MHC, albeit at the cost of utilizing more energy. Furthermore, researchers showed that calcium sensitivity was more attenuated in β-MHC than in α-MHC containing fibers [22]. Researchers' study revealed that the α-MHC isoform predominates in myosin heavy chain in sham-control rat hearts (90%), whereas, in comparison, the predominant phenotype is β-MHC in the BDL-cirrhotic heart [8]. This suggests that the shift from α-MHC to β-MHC in the cirrhotic heart plays an important role in the suppressed cardiac contractility of the cirrhotic heart [23].
Titin, a giant muscle protein, is mainly involved in ventricular diastolic function. In effect, its ‘spring-like’ elasticity is responsible for much of the passive mechanical recoil of early diastole. Kellermayer et al. demonstrated that titin abnormalities are associated with dilated cardiomyopathy. Researchers' group did not find any direct titin structural abnormalities in cirrhotic rat hearts. However, researchers showed that a titin modulator, protein kinase A, is decreased, which may contribute to abnormal titin function and thus reduced diastolic compliance in the cirrhotic ventricle [24].
Another abnormality of the intracellular calcium handling system is the calcium leakage from the SR. Calcium leakage reduces cardiac contractility in heart failure [25]. Researchers examined the root mean square value of sarcomere length fluctuations (RMSSL) to evaluate the amount of spontaneous sarcomere length fluctuation during diastole and found that RMSSL is significantly higher in ventricular trabeculae from cirrhotic rat hearts at all stimulus rates compared with that from sham-control rats [8]. This implies that the leakage of calcium from the SR in cirrhotic cardiomyocytes is higher than that from sham controls. Following contraction, the calcium leaks during diastole. In normal conditions, the random opening of RyR gives rise to calcium sparks and the calcium wave is small. However, in conditions such as congestive heart failure, the probability of spontaneous diastolic opening of SR calcium channels is increased, which causes noticeable fluctuations of diastolic sarcomere length and also the propagation of contractile waves in the myocytes [26]. Researchers' study demonstrated the increased spontaneous fluctuations of sarcomere length during diastole in BDL trabeculae, which suggests that there are dispersions of RyR [8]. In addition, Obayashi et al. [25] reported that the decreased force of cardiac contractility is proportional to the amount of spontaneous activity in chronic heart failure in rats. Researchers' data indicate that spontaneous SR-calcium release contributes to the reduction in inotropism in cirrhotic hearts. In addition, spontaneous random diastolic calcium release hinders the synchronization of individual sarcomeres, which also decreases the contractile force of cirrhotic hearts.
In addition to cardiac contractile dysfunction, calcium leakage also results in arrhythmias [15]. Calcium leakage causes delayed after-depolarizations (DADs) of the cell membrane due to the activity of the sodium–calcium exchanger. One such arrhythmia is atrial fibrillation [27]. It has been documented that atrial fibrillation is the most common arrhythmia found in patients with cirrhosis, with a prevalence between 6.6% and 14.2% [28][29]. Furthermore, atrial fibrillation is the most common arrhythmia in the perioperative period of both cirrhotic and noncirrhotic patients undergoing major surgery such as liver transplantation [30]. Atrial fibrillation is related to a variety of adverse outcomes [31] such as stroke, acute kidney injury, and in-hospital mortality. Understanding the role of calcium leakage in atrial fibrillation may eventually lead to new strategies to treat/prevent this arrhythmia in patients with cirrhosis.
References
- Yoon, K.T.; Liu, H.; Lee, S.S. Cirrhotic Cardiomyopathy. Curr. Gastroenterol. Rep. 2020, 22, 45.
- Ma, Z.; Lee, S.S. Cirrhotic cardiomyopathy: Getting to the heart of the matter. Hepatology 1996, 24, 451–459.
- Izzy, M.; VanWagner, L.B.; Lin, G.; Altieri, M.; Findlay, J.Y.; Oh, J.K.; Watt, K.D.; Lee, S.S.; Cirrhotic Cardiomyopathy, C. Redefining Cirrhotic Cardiomyopathy for the Modern Era. Hepatology 2020, 71, 334–345.
- Fialla, A.D.; Thiesson, H.C.; Bie, P.; Schaffalitzky de Muckadell, O.B.; Krag, A. Internal dysregulation of the renin system in patients with stable liver cirrhosis. Scand J. Clin. Lab. Investig. 2017, 77, 298–309.
- Uhlig, M.; Hein, M.; Habigt, M.A.; Tolba, R.H.; Braunschweig, T.; Helmedag, M.J.; Arici, M.; Theissen, A.; Klinkenberg, A.; Klinge, U.; et al. Cirrhotic Cardiomyopathy Following Bile Duct Ligation in Rats—A Matter of Time? Int. J. Mol Sci. 2023, 24, 8147.
- Nam, S.W.; Liu, H.; Wong, J.Z.; Feng, A.Y.; Chu, G.; Merchant, N.; Lee, S.S. Cardiomyocyte apoptosis contributes to pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated mice. Clin. Sci. 2014, 127, 519–526.
- Liu, L.; Liu, H.; Nam, S.W.; Lee, S.S. Protective effects of erythropoietin on cirrhotic cardiomyopathy in rats. Dig. Liver Dis. 2012, 44, 1012–1017.
- Honar, H.; Liu, H.; Zhang, M.L.; Glenn, T.K.; Ter Keurs, H.; Lee, S.S. Impaired myosin isoform shift and calcium transients contribute to cellular pathogenesis of rat cirrhotic cardiomyopathy. Liver Int. 2020, 40, 2808–2819.
- Ward, C.A.; Liu, H.; Lee, S.S. Altered cellular calcium regulatory systems in a rat model of cirrhotic cardiomyopathy. Gastroenterology 2001, 121, 1209–1218.
- Ward, C.A.; Ma, Z.; Lee, S.S.; Giles, W.R. Potassium currents in atrial and ventricular myocytes from a rat model of cirrhosis. Am. J. Physiol. 1997, 273, G537–G544.
- Kamp, T.J.; He, J.Q. L-type Ca2+ channels gaining respect in heart failure. Circ. Res. 2002, 91, 451–453.
- Valentim, M.A.; Brahmbhatt, A.N.; Tupling, A.R. Skeletal and cardiac muscle calcium transport regulation in health and disease. BioSci. Rep. 2022, 42, BSR20211997.
- Papa, A.; Kushner, J.; Marx, S.O. Adrenergic Regulation of Calcium Channels in the Heart. Annu. Rev. Physiol. 2022, 84, 285–306.
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195.
- Marks, A.R. Targeting ryanodine receptors to treat human diseases. J. Clin. Investig. 2023, 133, e162891.
- Moore, B. In Memory of Sidney Ringer : Some account of the Fundamental Discoveries of the Great Pioneer of the Bio-Chemistry of Crystallo-colloids in Living Cells. Biochem. J. 1911, 5, i.b3–xix.
- Mukherjee, R.; Hewett, K.W.; Walker, J.D.; Basler, C.G.; Spinale, F.G. Changes in L-type calcium channel abundance and function during the transition to pacing-induced congestive heart failure. Cardiovasc. Res. 1998, 37, 432–444.
- Kolstad, T.R.; van den Brink, J.; MacQuaide, N.; Lunde, P.K.; Frisk, M.; Aronsen, J.M.; Norden, E.S.; Cataliotti, A.; Sjaastad, I.; Sejersted, O.M.; et al. Ryanodine receptor dispersion disrupts Ca2+ release in failing cardiac myocytes. eLife 2018, 7, e39427.
- Shen, X.; van den Brink, J.; Bergan-Dahl, A.; Kolstad, T.R.; Norden, E.S.; Hou, Y.; Laasmaa, M.; Aguilar-Sanchez, Y.; Quick, A.P.; Espe, E.K.S.; et al. Prolonged beta-adrenergic stimulation disperses ryanodine receptor clusters in cardiomyocytes and has implications for heart failure. eLife 2022, 11, e77725.
- Shaldon, C.; Peacock, J.H.; Walker, R.M.; Palmer, D.B.; Badrick, F.E. The portal venous content of adrenaline and noradrenaline in portal hypertension. Lancet 1961, 1, 957–961.
- Henriksen, J.H.; Moller, S.; Ring-Larsen, H.; Christensen, N.J. The sympathetic nervous system in liver disease. J. Hepatol. 1998, 29, 328–341.
- Kawai, M.; Karam, T.S.; Michael, J.J.; Wang, L.; Chandra, M. Comparison of elementary steps of the cross-bridge cycle in rat papillary muscle fibers expressing alpha- and beta-myosin heavy chain with sinusoidal analysis. J. Muscle Res. Cell Motil. 2016, 37, 203–214.
- Liu, H.; Yoon, K.T.; Zhang, J.; Lee, S.S. Advances in cirrhotic cardiomyopathy. Curr. Opin. Gastroenterol. 2021, 37, 187–193.
- Glenn, T.K.; Honar, H.; Liu, H.; ter Keurs, H.E.; Lee, S.S. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J. Hepatol. 2011, 55, 1249–1255.
- Obayashi, M.; Xiao, B.; Stuyvers, B.D.; Davidoff, A.W.; Mei, J.; Chen, S.R.; ter Keurs, H.E. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc. Res. 2006, 69, 140–151.
- Davidoff, A.W.; Boyden, P.A.; Schwartz, K.; Michel, J.B.; Zhang, Y.M.; Obayashi, M.; Crabbe, D.; ter Keurs, H.E. Congestive heart failure after myocardial infarction in the rat: Cardiac force and spontaneous sarcomere activity. Ann. N. Y. Acad. Sci. 2004, 1015, 84–95.
- Vest, J.A.; Wehrens, X.H.; Reiken, S.R.; Lehnart, S.E.; Dobrev, D.; Chandra, P.; Danilo, P.; Ravens, U.; Rosen, M.R.; Marks, A.R. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005, 111, 2025–2032.
- Vandenberk, B.; Altieri, M.H.; Liu, H.; Raj, S.R.; Lee, S.S. Review article: Diagnosis, pathophysiology and management of atrial fibrillation in cirrhosis and portal hypertension. Aliment. Pharmacol. Ther. 2023, 57, 290–303.
- Gundling, F.; Schmidtler, F.; Zelihic, E.; Seidl, H.; Haller, B.; Ronel, J.; Loffler, N.; Schepp, W. Frequency of cardiac arrhythmia in patients with liver cirrhoses and evaluation of associated factors. Z. Gastroenterol. 2012, 50, 1149–1155.
- Altieri, M.H.; Liu, H.; Lee, S.S. Cardiovascular events after liver transplantation: MACE hurts. Rev. Cardiovasc. Med. 2022, 23, 91.
- Darrat, Y.H.; Smer, A.; Elayi, C.S.; Morales, G.X.; Alqahtani, F.; Alkhouli, M.; Catanzaro, J.; Shah, J.; Salih, M. Mortality and morbidity in patients with atrial fibrillation and liver cirrhosis. World J. Cardiol. 2020, 12, 342–350.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
778
Revisions:
3 times
(View History)
Update Date:
20 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No