 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karen W.L. Yee | -- | 3448 | 2023-07-17 04:22:10 | | | |
| 2 | Peter Tang | Meta information modification | 3448 | 2023-07-18 04:15:10 | | |
Video Upload Options
Myelodysplastic neoplasms (MDS) are a heterogenous group of clonal hematologic disorders characterized by morphologic dysplasia, ineffective hematopoiesis, and cytopenia. These three developments allow for more tailored therapeutic decision-making in view of the expanding treatment options in MDS. For patients with lower risk MDS, treatment is aimed at improving cytopenias, usually anemia. The approval of luspatercept and decitabine/cedazuridine have added on to the current armamentarium of erythropoietic stimulating agents and lenalidomide (for MDS with isolated deletion 5q). Several newer agents are being evaluated in phase 3 clinical trials for this group of patients, such as imetelstat and oral azacitidine.
1. Introduction
2. Classifications
|
WHO 2016 (4th ed) |
WHO 2022 (5th ed) |
ICC 2022 |
|---|---|---|
|
MDS with single lineage dysplasia (MDS-SLD) |
MDS with defining genetic abnormalities MDS with low blasts & isolated 5q deletion (MDS-5q) MDS with low blasts & SF3B1 mutation (MDS-SF3B1) a MDS with biallelic TP53 inactivation (MDS-biTP53) |
MDS with mutated SF3B1 |
|
MDS with multilineage dysplasia (MDS-MLD) |
MDS with del(5q) |
|
|
MDS with ring sideroblasts (MDS-RS) MDS-RS-SLD MDS-RS-MLD |
MDS with mutated TP53 |
|
|
MDS with isolated del(5q) |
MDS, not otherwise specified (MDS, NOS) MDS, NOS without dysplasia MDS, NOS with single lineage dysplasia MDS, NOS with multilineage dysplasia |
|
|
MDS with excess blasts (MDS-EB) MDS-EB-1 MDS-EB-2 |
MDS, morphologically defined MDS with low blasts (MDS-LB) MDS, hypoplastic (MDS-h) MDS with increased blasts (MDS-IB) MDS-IB1 MDS-IB2 MDS with fibrosis (MDS-f) |
MDS with excess blasts |
|
MDS, unclassifiable (MDS-U) |
MDS/AML b MDS/AML with mutated TP53 MDS/AML with myelodysplasia-related gene mutations MDS/AML with myelodysplasia-related cytogenetic abnormalities MDS/AML, NOS |
3. Risk Assessment
|
IPSS (Greenberg 1997) [9] |
IPSS-R (Greenberg 2012) [10] |
IPSS-M (Bernard 2022) [13] |
|
|---|---|---|---|
|
Includes CMML |
Yes a (if WBC ≤ 12 × 109/L) |
Yes b (if WBC ≤ 12 × 109/L) |
Yes c (if WBC < 13 × 109/L) |
|
Includes secondary/therapy-related MDS |
No |
No |
Yes d |
|
Includes previously treated patients |
No |
No |
Yes |
|
Sensitivity to degree of cytopenias |
Limited |
Anemia, thrombocytopenia & neutropenia |
Anemia & thrombocytopenia e |
|
Range of karyotypes |
3 categories |
5 categories |
5 categories |
|
Marrow blasts |
<30% a |
<30% b |
<20% |
|
Includes gene mutations |
No |
No |
Yes (31) |
|
Number of prognostic variables |
3 |
5 |
5 f |
|
Number of risk groups |
4 |
5 |
4 |
4. Myeloid Malignancies with Germline Predisposition
5. Therapeutic Options
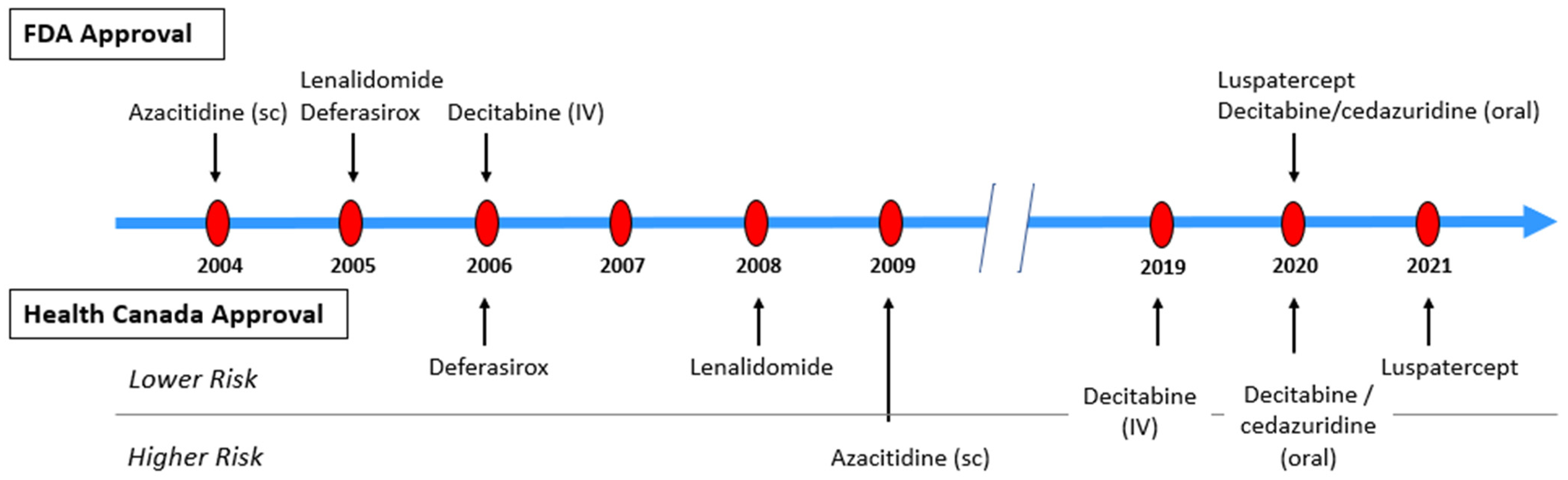
|
Drug |
Indication |
Regulatory Status |
Reference |
|---|---|---|---|
|
Azacitidine (AZA) |
for the treatment of patients with the following [FAB] MDS subtypes: refractory anemia or refractory anemia with ring sideroblasts (if accompanied by neutropenia or thrombocytopenia and requiring transfusions), refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia (CMML) |
FDA (2004) |
|
|
for the treatment of adult patients with (a) IPSS Intermediate-2 and High-risk MDS and (b) AML with 20–30% blasts and multi-lineage dysplasia, according to the WHO classification a,b |
FDA (expanded 2008); EMA (2008); HC (2009) |
Fenaux 2009 [39] |
|
|
Lenalidomide (LEN) |
for the treatment of transfusion-dependent anemia in patients with IPSS Low or Intermediate-1 risk MDS with chromosome 5q deletion c |
FDA (Sub-part H 2005); EMA (2013); HC (2008) |
Fenaux 2011 [40] |
|
Deferasirox (DFX) |
for use in treating chronic iron overload due to transfusional hemosiderosis in patients ≥ 2 years of age |
FDA (2005); EMA (2006) |
Shashaty 2006; Cappellini 2006; Cappellini 2011 |
|
for the management of chronic iron overload in patients with transfusion-dependent anemias aged ≥6 years and in patients aged 2 to 5 who cannot be adequately treated with deferoxamine |
HC (2006) |
||
|
Decitabine (DEC) |
for the treatment of adult patients with de novo or secondary MDS, untreated or previously treated with chemotherapy, including the following: (a) IPSS Intermediate-1, intermediate-2 and high-risk International Prognostic Scoring System (IPSS) groups and (b) all French-American-British (FAB) subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and CMML) a |
FDA (2006); HC (2019) |
Kantarjian 2006 [44] |
|
Decitabine/cedazuridine (DEC-C) |
for the treatment of adult patients with de novo or secondary MDS, untreated or previously treated with chemotherapy, including the following: (a) IPSS Intermediate-1, intermediate-2 and high-risk International Prognostic Scoring System (IPSS) groups and (b) all French-American-British (FAB) subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, and CMML) |
FDA (2020); HC (2020) |
|
|
Luspatercept |
for the treatment of anemia failing an ESA and requiring ≥2 RBC units over 8 weeks in adult patients with [IPSS-R] very low- to intermediate-risk MDS with ring sideroblasts (MDS-RS) d |
FDA (2020); EMA (2020); HC (2021) |
Fenaux 2020 [47] |
EMA—European Medicine Agencies; FDA—US Food and Drug Administration; HC—Health Canada. a HC approval only if patients are not considered candidates for HCT; b EMA approval only if patients are not considered candidates for HCT; c EMA approval only when other therapeutic options are insufficient or inadequate; d FDA approval also for patients with myelodysplastic/myeloproliferative neo-plasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T).
6. Newer Agents in Later Stages of Development
6.1. Imetelstat
6.2. Roxadustat
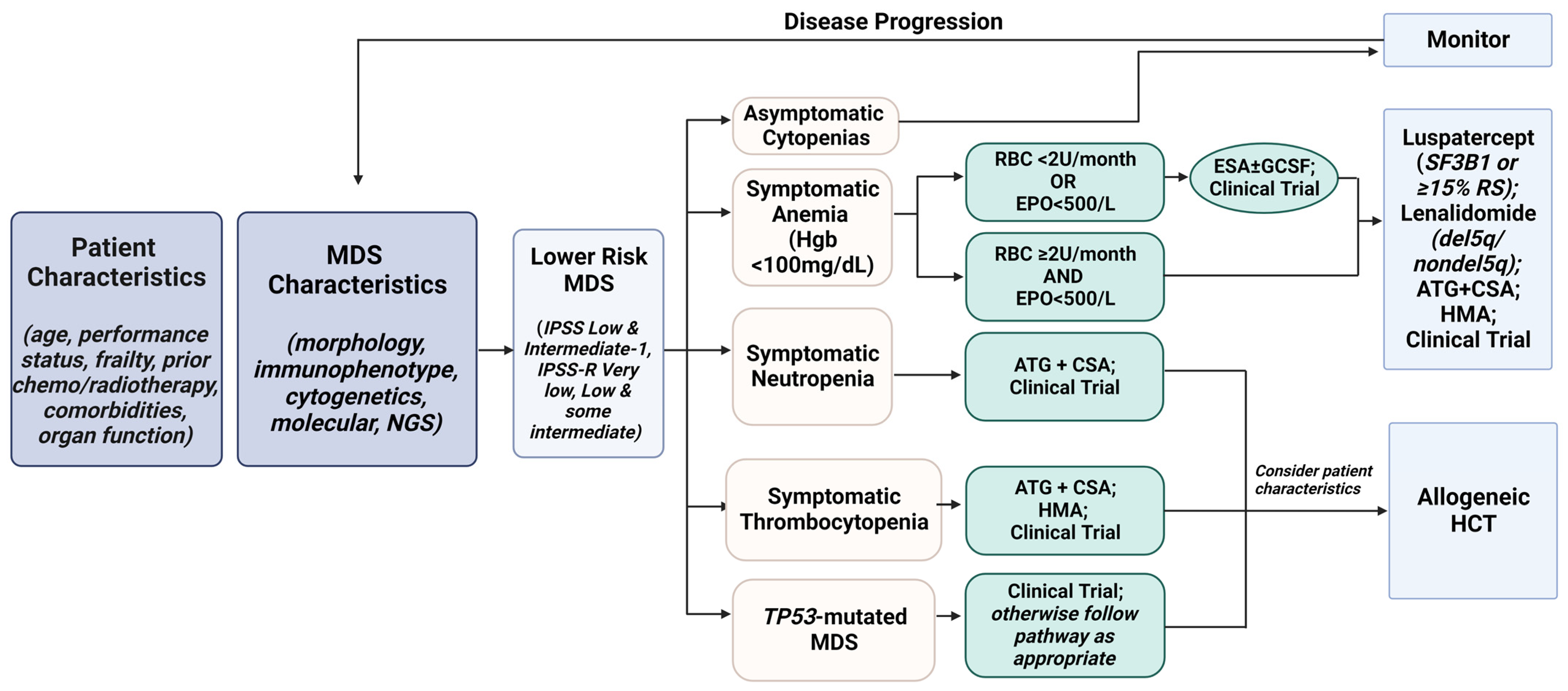
7. Treatment Algorithm for Lower-Risk MDS
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719.
- Board, W.C.o.T.E. Haematolymphoid Tumours , 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2022; Volume 11, Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 7 May 2023).
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951.
- Siebert, R.; Schuh, A.; Ott, G.; Cree, I.A.; Du, M.Q.; Ferry, J.; Hochhaus, A.; Naresh, K.N.; Solary, E.; Khoury, J.D. Response to the Comments from the Groupe Francophone de Cytogenetique Hematologique (GFCH) on the 5th edition of the World Health Organization classification of haematolymphoid tumors. Leukemia 2023, 37, 1170–1172.
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556.
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758.
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088.
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465.
- Pfeilstocker, M.; Tuechler, H.; Sanz, G.; Schanz, J.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Time-dependent changes in mortality and transformation risk in MDS. Blood 2016, 128, 902–910.
- Benton, C.B.; Khan, M.; Sallman, D.; Nazha, A.; Nogueras Gonzalez, G.M.; Piao, J.; Ning, J.; Aung, F.; Al Ali, N.; Jabbour, E.; et al. Prognosis of patients with intermediate risk IPSS-R myelodysplastic syndrome indicates variable outcomes and need for models beyond IPSS-R. Am. J. Hematol. 2018, 93, 1245–1253.
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for myelodysplastic syndromes. NEJM Evid. 2022, 1, EVIDoa2200008.
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377.
- Keel, S.B.; Scott, A.; Sanchez-Bonilla, M.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.C.; Walsh, T.; Shimamura, A. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica 2016, 101, 1343–1350.
- Feurstein, S.; Churpek, J.E.; Walsh, T.; Keel, S.; Hakkarainen, M.; Schroeder, T.; Germing, U.; Geyh, S.; Heuser, M.; Thol, F.; et al. Germline variants drive myelodysplastic syndrome in young adults. Leukemia 2021, 35, 2439–2444.
- Feurstein, S.; Trottier, A.M.; Estrada-Merly, N.; Pozsgai, M.; McNeely, K.; Drazer, M.W.; Ruhle, B.; Sadera, K.; Koppayi, A.L.; Scott, B.L.; et al. Germ line predisposition variants occur in myelodysplastic syndrome patients of all ages. Blood 2022, 140, 2533–2548.
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: Current strategies and challenges. Br. J. Haematol. 2022, 196, 1293–1310.
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247.
- NCCN. Clinical Practice Guidelines in Oncology—Myelodysplastic Syndromes V.1.2023; National Comprehensive Cancer Network, Inc.: Plymouth Meeting, PA, USA, 2023; Available online: https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf (accessed on 7 May 2023).
- Ansar, S.; Malcolmson, J.; Farncombe, K.M.; Yee, K.; Kim, R.H.; Sibai, H. Clinical implementation of genetic testing in adults for hereditary hematologic malignancy syndromes. Genet. Med. 2022, 24, 2367–2379.
- Dayyani, F.; Conley, A.P.; Strom, S.S.; Stevenson, W.; Cortes, J.E.; Borthakur, G.; Faderl, S.; O’Brien, S.; Pierce, S.; Kantarjian, H.; et al. Cause of death in patients with lower-risk myelodysplastic syndrome. Cancer 2010, 116, 2174–2179.
- Gyan, E.; Andrieu, V.; Sanna, A.; Caille, A.; Schemenau, J.; Sudaka, I.; Siguret, V.; Malet, M.; Park, S.; Bordessoule, D.; et al. Myelodysplastic syndromes with single neutropenia or thrombocytopenia are rarely refractory cytopenias with unilineage dysplasia by World Health Organization 2008 criteria and have favourable prognosis. Br. J. Haematol. 2016, 175, 975–979.
- Toma, A.; Fenaux, P.; Dreyfus, F.; Cordonnier, C. Infections in myelodysplastic syndromes. Haematologica 2012, 97, 1459–1470.
- Kantarjian, H.; Giles, F.; List, A.; Lyons, R.; Sekeres, M.A.; Pierce, S.; Deuson, R.; Leveque, J. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007, 109, 1705–1714.
- Hutzschenreuter, F.; Monsef, I.; Kreuzer, K.A.; Engert, A.; Skoetz, N. Granulocyte and granulocyte-macrophage colony stimulating factors for newly diagnosed patients with myelodysplastic syndromes. Cochrane Database Syst. Rev. 2016, 2, CD009310.
- Giagounidis, A.; Mufti, G.J.; Fenaux, P.; Sekeres, M.A.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Hu, K.; et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1-risk myelodysplastic syndrome and thrombocytopenia. Cancer 2014, 120, 1838–1846.
- Oliva, E.N.; Riva, M.; Niscola, P.; Santini, V.; Breccia, M.; Giai, V.; Poloni, A.; Patriarca, A.; Crisa, E.; Capodanno, I.; et al. Eltrombopag for low-risk myelodysplastic syndromes with thrombocytopenia: Interim results of a phase-II, randomized, placebo-controlled clinical trial (EQOL-MDS). J. Clin. Oncol. 2023, 1–11.
- Kantarjian, H.M.; Fenaux, P.; Sekeres, M.A.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Carpenter, N.; Mehta, B.; et al. Long-term follow-up for up to 5 years on the risk of leukaemic progression in thrombocytopenic patients with lower-risk myelodysplastic syndromes treated with romiplostim or placebo in a randomised double-blind trial. Lancet Haematol. 2018, 5, e117–e126.
- Kubasch, A.S.; Giagounidis, A.; Metzgeroth, G.; Jonasova, A.; Herbst, R.; Diaz, J.M.T.; De Renzis, B.; Gotze, K.S.; Huetter-Kroenke, M.L.; Gourin, M.P.; et al. Prospective validation of a biomarker-driven response prediction model to romiplostim in lower-risk myelodysplastic neoplasms—Results of the EUROPE trial by EMSCO. Leukemia 2022, 36, 2519–2527.
- Platzbecker, U.; Hofbauer, L.C.; Ehninger, G.; Holig, K. The clinical, quality of life, and economic consequences of chronic anemia and transfusion support in patients with myelodysplastic syndromes. Leuk. Res. 2012, 36, 525–536.
- Santini, V. Treatment of low-risk myelodysplastic syndromes. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 462–469.
- Germing, U.; Lauseker, M.; Hildebrandt, B.; Symeonidis, A.; Cermak, J.; Fenaux, P.; Kelaidi, C.; Pfeilstocker, M.; Nosslinger, T.; Sekeres, M.; et al. Survival, prognostic factors and rates of leukemic transformation in 381 untreated patients with MDS and del(5q): A multicenter study. Leukemia 2012, 26, 1286–1292.
- Goldberg, S.L.; Chen, E.; Corral, M.; Guo, A.; Mody-Patel, N.; Pecora, A.L.; Laouri, M. Incidence and clinical complications of myelodysplastic syndromes among United States Medicare beneficiaries. J. Clin. Oncol. 2010, 28, 2847–2852.
- Schafer, A.I.; Cheron, R.G.; Dluhy, R.; Cooper, B.; Gleason, R.E.; Soeldner, J.S.; Bunn, H.F. Clinical consequences of acquired transfusional iron overload in adults. N. Engl. J. Med. 1981, 304, 319–324.
- Silverman, L.R.; McKenzie, D.R.; Peterson, B.L.; Holland, J.F.; Backstrom, J.T.; Beach, C.L.; Larson, R.A.; Cancer and Leukemia Group, B. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: Studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J. Clin. Oncol. 2006, 24, 3895–3903.
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440.
- Kornblith, A.B.; Herndon, J.E., II; Silverman, L.R.; Demakos, E.P.; Odchimar-Reissig, R.; Holland, J.F.; Powell, B.L.; DeCastro, C.; Ellerton, J.; Larson, R.A.; et al. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2002, 20, 2441–2452.
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232.
- Fenaux, P.; Giagounidis, A.; Selleslag, D.; Beyne-Rauzy, O.; Mufti, G.; Mittelman, M.; Muus, P.; Te Boekhorst, P.; Sanz, G.; Del Canizo, C.; et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011, 118, 3765–3776.
- Shashaty, G.; Frankewich, R.; Chakraborti, T.; Choudary, J.; Al-Fayoumi, S.; Kacuba, A.; Castillo, S.; Robie-Suh, K.; Rieves, D.; Weiss, K.; et al. Deferasirox for the treatment of chronic iron overload in transfusional hemosiderosis. Oncology 2006, 20, 1799–1806, 1811; discussion 1811–1813, 1817.
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462.
- Cappellini, M.D.; Bejaoui, M.; Agaoglu, L.; Canatan, D.; Capra, M.; Cohen, A.; Drelichman, G.; Economou, M.; Fattoum, S.; Kattamis, A.; et al. Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: Efficacy and safety during 5 years’ follow-up. Blood 2011, 118, 884–893.
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803.
- Garcia-Manero, G.; McCloskey, J.; Griffiths, E.A.; Yee, K.W.L.; Zeidan, A.M.; Al-Kali, A.; Dao, K.H.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; et al. Pharmacokinetic exposure equivalence and preliminary efficacy and safety from a randomized cross over phase 3 study (ASCERTAIN study) of an oral hypomethylating agent ASTX727 (cedazuridine/decitabine) compared to IV decitabine. Blood 2019, 134 (Suppl. S1), 846.
- Savona, M.R.; McCloskey, J.K.; Griffiths, E.A.; Yee, K.W.L.; Al-Kali, A.; Zeidan, A.M.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; Keating, M.M.; et al. Clinical efficacy and safety of oral decitabine/cedazuridine in 133 patients with myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemia (CMML). Blood 2020, 136 (Suppl. S1), 37–38.
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Diez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med. 2020, 382, 140–151.
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat achieves meaningful and durable transfusion independence in high transfusion-burden patients with lower-risk myelodysplastic syndromes in a phase II study. J. Clin. Oncol. 2021, 39, 48–56.
- Platzbecker, U.; Komrokji, R.S.; Fenaux, P.; Zeidan, A.M.; Sekeres, M.A.; Savona, M.R.; Madanat, Y.F.; Santini, V.; Van Eygen, K.; Raza, A.; et al. Imetelstat achieved prolonged, continuous transfusion independence (TI) in patients with heavily transfused non-del(5q) lower-risk myelodysplastic syndrome (LR-MDS) relapsed/refractory (R/R) to erythropoiesis stimulating agents (ESAs) within the IMerge phase 2 study. Blood 2022, 140 (Suppl. S1), 1106–1108.
- Geron Announces Positive Top-Line Results from IMerge Phase 3 Trial of Imetelstat in Lower Risk MDS. 2023. Available online: https://ir.geron.com/investors/press-releases/press-release-details/2023/Geron-Announces-Positive-Top-Line-Results-from-IMerge-Phase-3-Trial-of-Imetelstat-in-Lower-Risk-MDS/default.aspx (accessed on 7 May 2023).
- Besarab, A.; Provenzano, R.; Hertel, J.; Zabaneh, R.; Klaus, S.J.; Lee, T.; Leong, R.; Hemmerich, S.; Yu, K.H.; Neff, T.B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol. Dial. Transplant. 2015, 30, 1665–1673.
- Besarab, A.; Chernyavskaya, E.; Motylev, I.; Shutov, E.; Kumbar, L.M.; Gurevich, K.; Chan, D.T.; Leong, R.; Poole, L.; Zhong, M.; et al. Roxadustat (FG-4592): Correction of anemia in incident dialysis patients. J. Am. Soc. Nephrol. 2016, 27, 1225–1233.
- Henry, D.H.; Glaspy, J.; Harrup, R.; Mittelman, M.; Zhou, A.; Carraway, H.E.; Bradley, C.; Saha, G.; Modelska, K.; Bartels, P.; et al. Roxadustat for the treatment of anemia in patients with lower-risk myelodysplastic syndrome: Open-label, dose-selection, lead-in stage of a phase 3 study. Am. J. Hematol. 2022, 97, 174–184.
- Aprea Therapeutics Announces Results of Primary Endpoint from Phase 3 Trial of Eprenetapopt in TP53 Mutant Myelodysplastic Syndromes (MDS). 2020. Available online: https://ir.aprea.com/news-releases/news-release-details/aprea-therapeutics-announces-results-primary-endpoint-phase-3 (accessed on 7 May 2023).
- Zeidan, A.M.; Al Ali, N.H.; Padron, E.; Lancet, J.; List, A.; Komrokji, R.S. Lenalidomide treatment for lower risk nondeletion 5q myelodysplastic syndromes patients yields higher response rates when used before azacitidine. Clin. Lymphoma Myeloma Leuk. 2015, 15, 705–710.
- Zeidan, A.M.; Klink, A.J.; McGuire, M.; Feinberg, B. Treatment sequence of lenalidomide and hypomethylating agents and the impact on clinical outcomes for patients with myelodysplastic syndromes. Leuk. Lymphoma 2019, 60, 2050–2055.
- Komrokji, R.S.; Al Ali, N.; Ball, S.; Chan, O.; Kuykendall, A.; Sweet, K.; Lancet, J.E.; Padron, E.; Sallman, D.A. Luspatercept for treatment of lower risk myelodysplastic syndromes: Real world data replicates Medalist study results and confirms activity among hypomethylating agents and lenalidomide treated patients. Blood 2022, 140 (Suppl. S1), 4039–4041.


