Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jeffrey Yuchen Jian | -- | 1957 | 2023-07-14 17:11:30 | | | |
| 2 | Sirius Huang | Meta information modification | 1957 | 2023-07-17 04:24:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jian, J.Y.; Osheroff, N. Type II Topoisomerases. Encyclopedia. Available online: https://encyclopedia.pub/entry/46823 (accessed on 25 July 2026).
Jian JY, Osheroff N. Type II Topoisomerases. Encyclopedia. Available at: https://encyclopedia.pub/entry/46823. Accessed July 25, 2026.
Jian, Jeffrey Y., Neil Osheroff. "Type II Topoisomerases" Encyclopedia, https://encyclopedia.pub/entry/46823 (accessed July 25, 2026).
Jian, J.Y., & Osheroff, N. (2023, July 14). Type II Topoisomerases. In Encyclopedia. https://encyclopedia.pub/entry/46823
Jian, Jeffrey Y. and Neil Osheroff. "Type II Topoisomerases." Encyclopedia. Web. 14 July, 2023.
Copy Citation
Type II topoisomerases are essential enzymes that modulate the topological state of DNA supercoiling in all living organisms. These enzymes alter DNA topology by performing double-stranded passage reactions on over- or underwound DNA substrates. This strand passage reaction generates a transient covalent enzyme–cleaved DNA structure known as the cleavage complex.
DNA topoisomerase
DNA topology
supercoil handedness
type II topoisomerase
1. Topoisomerases
In order to maintain appropriate levels of DNA supercoiling and remove knots and tangles from the genome, cells encode enzymes known as topoisomerases [1][2][3][4][5][6]. These enzymes are ubiquitous to all domains of life and are necessary for cellular survival. All topoisomerases modulate the topological state of the genome through the creation of transient breaks in the DNA sugar–phosphate backbone. Broadly, there are two classes of topoisomerases, and they are both defined by the number of DNA strands they cleave per enzyme reaction cycle [1][2][4][5][7]. Type I topoisomerases generate a single-stranded break, or “nick”, in the double helix [2][4][5]. In contrast, type II topoisomerases create a double-stranded break in the genetic material [1][2][3][4][5][7][8].
2. Type II Topoisomerases
There are two subclasses of type II topoisomerases: type IIA and type IIB. To date, functional type IIB enzymes have only been identified in plants and archaea and will not be discussed further [5][9][10].
The first type IIA enzyme, bacterial DNA gyrase, was discovered in 1976 [11]. Bacterial topoisomerase IV was later identified in 1990 [12]. Most bacterial species encode gyrase and topoisomerase IV [5][13]. However, a few species, such as Mycobacterium tuberculosis, encode only a single type II topoisomerase, gyrase, which can presumably perform the cellular functions of both type II enzymes [1][3][6][14][15].
The first eukaryotic type II enzyme was identified in Drosophila in 1980 [16]. Drosophila and other invertebrates, as well as lower eukaryotes, such as yeast, encode only one type II enzyme, topoisomerase II. In contrast, vertebrates, such as humans, express two forms of the type II enzyme: topoisomerase IIα and topoisomerase IIβ [1][2][4][5][8][17][18]. Human topoisomerase IIα and topoisomerase IIβ were identified in 1988 [19] and 1989 [20][21], respectively.
3. Type II Topoisomerase Domain Structures
Bacterial type II topoisomerases are heterotetrameric in structure (A2B2; Figure 1). The founding type II enzyme, gyrase, is comprised of two distinct subunits: GyrA and GyrB. Like gyrase, topoisomerase IV is a heterotetramer that is composed of two separate subunits: ParC and ParE (which are homologous to GyrA and GyrB, respectively) in Gram-negative species and the corresponding GrlA and GrlB subunits in Gram-positive species (Figure 1) [12][22].
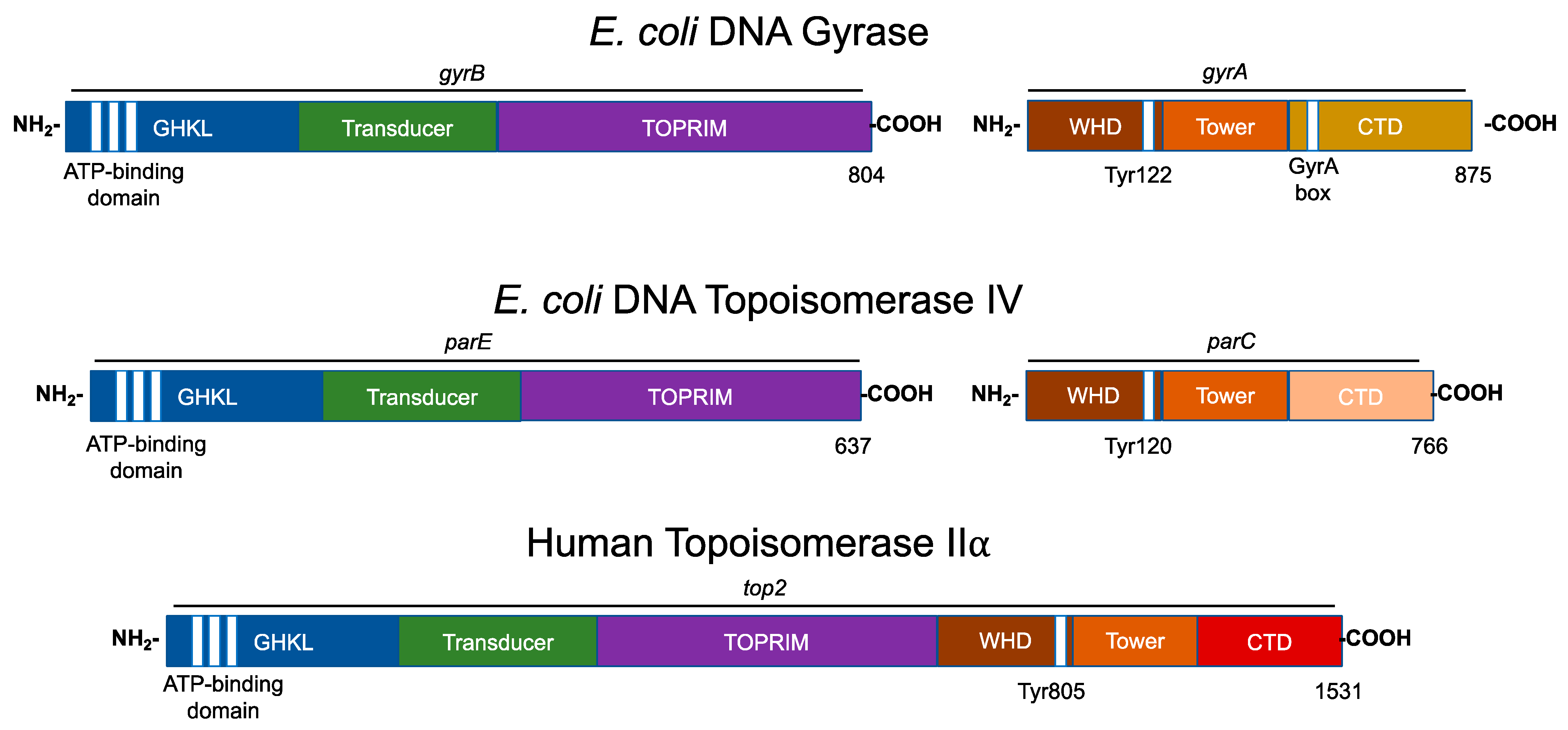
Figure 1. Domain structures of type II topoisomerases. The domain structures of three type II topoisomerases, bacterial (Escherichia coli) gyrase and topoisomerase IV, and human topoisomerase IIα are shown. Regions of homology among the enzymes are indicated by colors. The N-terminal (i.e., GyrB) homology domains contain the regions responsible for ATP binding and hydrolysis (GHKL, blue). The vertical white stripes represent the three conserved motifs that define the ATP-binding domain. The N-terminal domain also contains the binding site for divalent metal ions (TOPRIM, purple). The central (i.e., GyrA) region (WHD, brown) contains the active site tyrosyl residue that forms the covalent bond with DNA during scission. For bacterial gyrase, the variable C-terminal domain (gyrase, gold; topoisomerase IV, pink) contains the “GyrA box” that is necessary for the wrapping mechanism. For human topoisomerase IIα, the C-terminal homology domain (CTD, red) contains nuclear localization sequences (NLS) and phosphorylation sites (PO4). The active site tyrosine residue is indicated for each enzyme.
Eukaryotic type II topoisomerases are homologous to the bacterial type II enzymes [1][2][3][4][5][23]. However, the two bacterial subunits have fused into a single polypeptide in the eukaryotic type II topoisomerases (Figure 1) [1][2][3][4][5][23].
All known type II topoisomerases share several common structural features across three regions. Using DNA gyrase as the model, the N-terminus is located in GyrB, the catalytic core spans portions of GyrB and GyrA, and the C-terminus is located in GyrA (Figure 1) [1][2][3][4][5][23].
The N-terminal region contains the N-gate, where the DNA enters the enzyme. This portion of the molecule includes the ATPase active site, also known as the GHKL (DNA gyrase, Hsp90, bacterial CheA-family histidine kinases, and MutL; Figure 1, blue) domain. The GHKL domain contains an ATP-binding region that is formed from an eight-stranded antiparallel beta sheet surrounded by alpha helices [24][25]. The N-terminal region also contains the transducer domain (Figure 1, green), which relays ATP binding/hydrolysis information to the catalytic core [26][27]. The binding of ATP induces the dimerization of the N-terminal region, which shifts the N-gate into a closed conformation. The bound ATP interacts with a lysine residue in the transducer domain and subsequently facilitates rotation between the GHKL and transducer domains [9][25].
The catalytic core contains the topoisomerase/primase (TOPRIM; Figure 1, purple) domain, which coordinates the active site divalent cations, the winged-helix domain (WHD; Figure 1, brown), which contains the active site tyrosine residue, and the tower domain (Figure 1; orange), which maintains polar and electrostatic interactions with the DNA substrate [26][28][29].
The TOPRIM domain is necessary for the transesterification reaction between the scissile phosphate of the DNA backbone and active site tyrosine residue [25][28]. The active site divalent cation is held by an aspartate-any residue-aspartate (DxD) motif and a glutamate residue that can act as a general acid–base moiety [30][31]. The DxD motif and its coordinate divalent cation in the TOPRIM domain, along with the active site tyrosine of the WHD, enable the formation of the two transient cuts of the DNA backbone via a non-canonical two-metal ion mechanism [1][29][32][33].
The WHD is able to bind DNA and also contains the active site tyrosine residue, which is responsible for the nucleophilic attack on the scissile phosphate of the DNA double helical backbone and the formation of the transient topoisomerase–DNA covalent bond [23][26].
The tower domain functions in DNA bending. This domain contains a beta sheet that can interact with one of the captured DNA double helices (the gate or G-segment, to be discussed later), bending the DNA segment to promote cleavage [34][35][36]. The presence of a conserved, invariant isoleucine residue has been found to intercalate between two base pairs of the G-segment, inducing a ~150° bend [34][37]. The deletion or mutation of this isoleucine interferes with proper DNA bending, the subsequent cleavage, and the relaxation of supercoiled DNA [34][37].
The sequence of the C-terminal domain varies considerably between species, but it is characterized by the presence of charged amino acid residues [5][38]. In gyrase, this region contains a seven-amino acid motif known as the GyrA box (Figure 1, gold) [3][13][25][39][40]. The GyrA box is located within a six-blade beta pinwheel in the C-terminal domain, and it uniquely allows for the wrapping of the DNA substrate to introduce (−)SCs [3][13][25][39][40][41].
In comparison to gyrase, the C-terminal domain of topoisomerase IV does not contain the structure necessary to wrap and supercoil DNA (Figure 1, pink). Rather, topoisomerase IV contains a “broken” five- (not six) blade beta pinwheel and lacks a GyrA box [24][42][43][44]. Remnants of the canonical GyrA motif have been found in each of its pinwheel “blades” [42][43]. Nonetheless, the C-terminal domain of topoisomerase IV contains positively charged moieties on its outer surface, suggesting a role in binding DNA [24].
The C-termini of eukaryotic type II topoisomerases also contain the remnants of highly charged pinwheel blades but are inherently disordered in the absence of DNA (Figure 1, red) [45][46]. This portion of the eukaryotic enzyme also contains nuclear localization sequences and sites for posttranslational modifications such as phosphorylation and SUMOylation [4][23][25]. For the IIα isoform, these modifications enable the enzyme to be concentrated at centromeres during mitosis [45][47].
4. Catalytic Cycle of Type II Topoisomerases
All type II topoisomerases undergo similar catalytic cycles. These enzymes function by forming a transient double-stranded DNA break and modulate the topological state of DNA by a double-stranded passage reaction (Figure 2) [1][2][3][4][5][8][25]. The enzyme begins its catalytic cycle by capturing a segment of intact DNA through the opened N-terminal region (N-gate, gray) of the enzyme (Step 1). This first segment will be cut by the enzyme and is known as the “gate” or G-segment. The segment that is captured second and eventually transported through the transiently cleaved G-segment is known as the “transport” or T-segment. In the presence of two divalent cations, such as Mg2+, and in coordination with the TOPRIM domain, the G-segment is assessed for bendability (Step 2) [35]. DNA sequences that can be bent are distorted to an angle of ~150° and can be used as the site for scission [34][35][36][37].
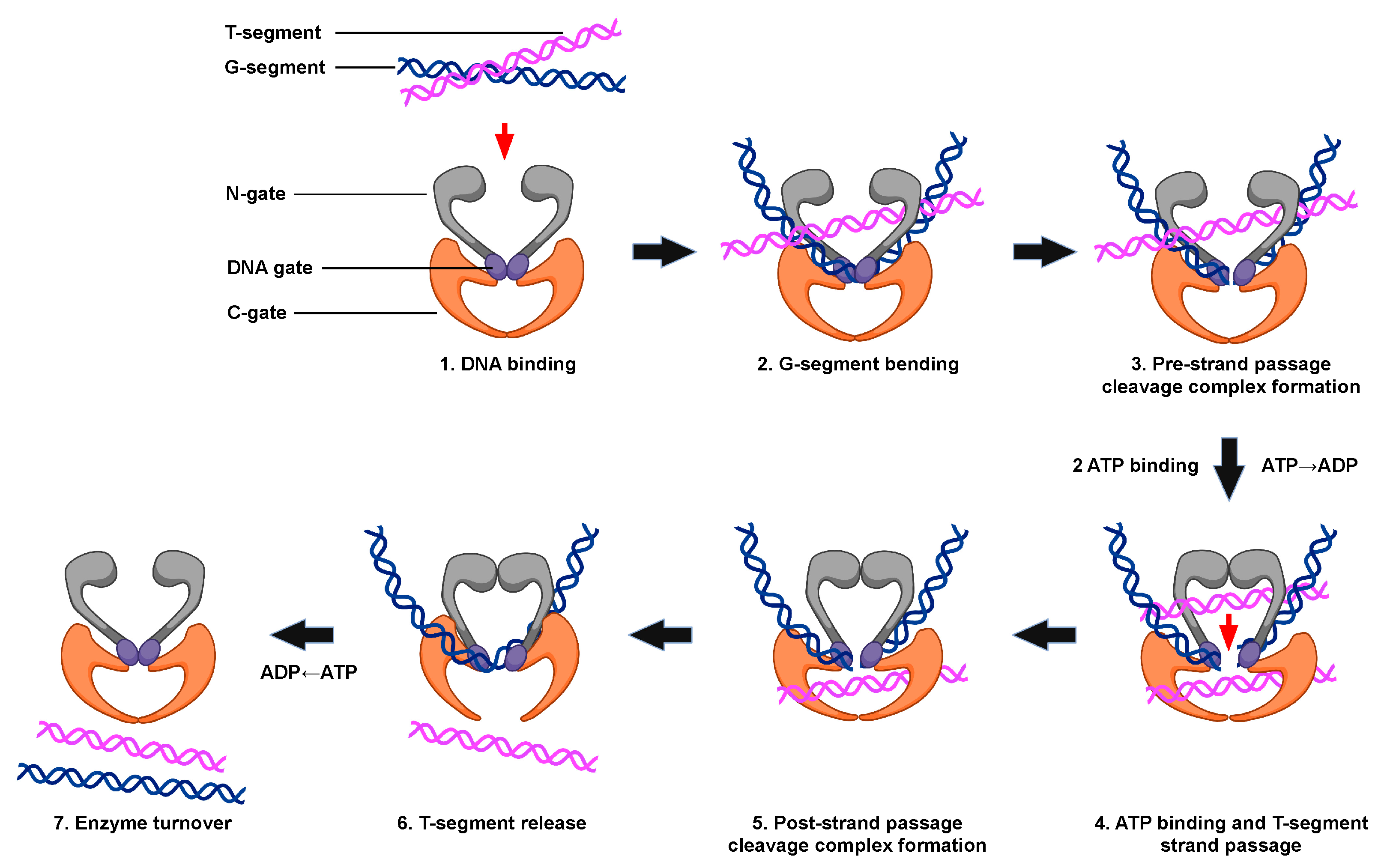
Figure 2. The catalytic cycle of type II topoisomerases. The double-stranded DNA passage reaction of type II topoisomerases can be separated into discrete steps. (1) Type II enzyme binding to two intact segments of DNA: the gate, or G-segment (blue, the first segment bound) and transport, or T-segment (purple, the second segment bound). (2) Bending of the G-segment to assess for sites of DNA cleavage. (3) Double-stranded DNA cleavage of the G-segment (i.e., formation of the pre-strand cleavage complex). (4) Binding of two ATP molecules, which triggers the closing of the N-gate, opening of the DNA gate, and the passage of the T-segment through the DNA gate. Strand passage occurs more rapidly if one of the two ATP molecules is hydrolyzed. (5) Formation of the post-strand passage cleavage complex. (6) Religation of the cleaved G-segment and release of the T-segment through the C-gate of the protein. (7) ATP hydrolysis, which triggers enzyme turnover and the regeneration of the enzyme to initiate a new round of catalysis. Created with BioRender.com.
Both strands of the bent G-segment are then cleaved via a nucleophilic attack by the two active site tyrosine residues on the phosphate backbone of the double helix (Step 3). DNA cleavage is initiated when a general base, which is believed to be a conserved histidine residue, deprotonates the hydroxyl group of the active site tyrosine, allowing the oxyanion to attack the scissile phosphate. Two divalent cation molecules, such as magnesium (i.e., Mg2+), are necessary for this nucleophilic attack [1][3][4][5][48]. Type II topoisomerases use a non-canonical two-metal ion mechanism [29][48]. The presence of one divalent cation enables interaction with the bridging 5′-oxygen molecule of the scissile bond and speeds up rates of enzyme-mediated cleavage at the first cut site. Once the first DNA strand is cut, the second strand is cleaved ~20-fold faster [49]. The resulting enzyme-cleaved DNA complex is a transient structure that has the enzyme covalently bound to the scissile 5′-phosphate of the double helical backbone.
To maintain the bond energy of the sugar–phosphate backbone as well as genomic integrity during the double-stranded DNA cleavage process, the type II enzyme forms covalent bonds between the two active site tyrosine residues and the newly generated 5′-phosphate groups of the DNA backbone, generating a phosphotyrosyl linkage and a four-base DNA overhang [1][3][4][8][25]. The transiently cleaved, covalently linked enzyme–DNA structure that is formed is known as the pre-strand passage “cleavage complex” [1][3][4][8]. The formation of the cleavage complex during enzyme catalysis is tightly regulated to prevent the generation of permanent DNA breaks or the disruption of genomic integrity [1][3][4][5].
When ATP enters the enzyme–DNA complex is not precisely known. This high-energy cofactor is not required for either DNA cleavage or religation. However, upon the binding of two ATP molecules, the N-gate is closed, triggering a conformational change in the enzyme that translocates the T-segment through the transient opening in the DNA (i.e., DNA “gate”, Step 4). Although hydrolysis of the high-energy cofactor is not necessary for this strand passage event to occur, this step proceeds faster if one of the two bound ATP molecules is hydrolyzed [50].
After strand passage, a second, post-strand passage, cleavage complex is formed (Step 5). The type II enzyme then religates the cleaved DNA to regenerate the intact DNA double helix. DNA religation is initiated when a general acid removes the hydrogen from the 3′-terminal hydroxyl group [6][48]. Another nucleophilic attack is then initiated on the phosphotyrosyl bond, regenerating the intact DNA double helical backbone and the enzyme active site. The T-segment is then released from the protein (Step 6). The hydrolysis of a second ATP molecule occurs, resetting the type II enzyme conformation and allowing for enzyme turnover during the next cycle of catalysis (Step 7).
References
- Deweese, J.E.; Osheroff, M.A.; Osheroff, N. DNA topology and topoisomerases: Teaching a “knotty” subject. Biochem. Mol. Biol. Educ. 2008, 37, 2–10.
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721.
- Ashley, R.E.; Osheroff, N. Regulation of DNA topology by topoisomerases: Mathematics at the molecular level. In Knots, Low-dimensional Topology and Applications; Adams, C.C., Gordon, C.M., Jones, V.F.R., Kauffman, L.H., Lambropoulou, S., Millett, K., Przytycki, J.H., Ricca, R., Sazdanovic, R., Eds.; Springer Proceedings in Mathematics & Statistics; Springer: New York, NY, USA, 2019; Volume 284, pp. 411–433.
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748.
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell. Biol. 2011, 12, 827–841.
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170.
- Bates, A.D.; Maxwell, A. DNA Topology; Oxford University Press: New York, NY, USA, 2005; p. 220.
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II poisons: Converting essential enzymes into molecular scissors. Biochemistry 2021, 60, 1630–1641.
- Corbett, K.D.; Berger, J.M. Structure of the topoisomerase VI-B subunit: Implications for type II topoisomerase mechanism and evolution. EMBO J. 2003, 22, 151–163.
- Forterre, P.; Gadelle, D. Phylogenomics of DNA topoisomerases: Their origin and putative roles in the emergence of modern organisms. Nucleic Acids Res. 2009, 37, 679–692.
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Nash, H.A. DNA gyrase: An enzyme that introduces superhelical turns into DNA. Proc. Natl. Acad. Sci. USA 1976, 73, 3872–3876.
- Kato, J.; Nishimura, Y.; Imamura, R.; Niki, H.; Hiraga, S.; Suzuki, H. New topoisomerase essential for chromosome segregation in E. coli. Cell 1990, 63, 393–404.
- Sissi, C.; Palumbo, M. In front of and behind the replication fork: Bacterial type IIA topoisomerases. Cell. Mol. Life. Sci. 2010, 67, 2001–2024.
- Forterre, P.; Gribaldo, S.; Gadelle, D.; Serre, M.C. Origin and evolution of DNA topoisomerases. Biochimie 2007, 89, 427–446.
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544.
- Hsieh, T.; Brutlag, D.L. ATP-dependent DNA topoisomerase from D. melanogaster reversibly catenates duplex DNA rings. Cell 1980, 21, 115–125.
- Liu, Z.; Deibler, R.W.; Chan, H.S.; Zechiedrich, L. The why and how of DNA unlinking. Nucleic Acids Res. 2009, 37, 661–671.
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The first thirty years. Int. J. Mol. Sci. 2018, 19, 2765.
- Tsai-Pflugfelder, M.; Liu, L.F.; Liu, A.A.; Tewey, K.M.; Whang-Peng, J.; Knutsen, T.; Huebner, K.; Croce, C.M.; Wang, J.C. Cloning and sequencing of cDNA encoding human DNA topoisomerase II and localization of the gene to chromosome region 17q21-22. Proc. Natl. Acad. Sci. USA 1988, 85, 7177–7181.
- Drake, F.H.; Hofmann, G.A.; Bartus, H.F.; Mattern, M.R.; Crooke, S.T.; Mirabelli, C.K. Biochemical and pharmacological properties of p170 and p180 forms of topoisomerase II. Biochemistry 1989, 28, 8154–8160.
- Chung, T.D.; Drake, F.H.; Tan, K.B.; Per, S.R.; Crooke, S.T.; Mirabelli, C.K. Characterization and immunological identification of cDNA clones encoding two human DNA topoisomerase II isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 9431–9435.
- Kato, J.; Suzuki, H.; Ikeda, H. Purification and characterization of DNA topoisomerase IV in Escherichia coli. J. Biol. Chem. 1992, 267, 25676–25684.
- Dalvie, E.D.; Osheroff, N. DNA topoisomerases: Type II. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Oxford, UK, 2021; pp. 479–486.
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118.
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. Bioessays 2021, 43, 2000286.
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase IIα: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124.
- Bjergbaek, L.; Kingma, P.; Nielsen, I.S.; Wang, Y.; Westergaard, O.; Osheroff, N.; Andersen, A.H. Communication between the ATPase and cleavage/religation domains of human topoisomerase IIα. J. Biol. Chem. 2000, 275, 13041–13048.
- Chang, C.C.; Wang, Y.R.; Chen, S.F.; Wu, C.C.; Chan, N.L. New insights into DNA-binding by type IIA topoisomerases. Curr. Opin. Struct. Biol. 2013, 23, 125–133.
- Schmidt, B.H.; Burgin, A.B.; Deweese, J.E.; Osheroff, N.; Berger, J.M. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 2010, 465, 641–644.
- Aravind, L.; Leipe, D.D.; Koonin, E.V. Toprim—A conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998, 26, 4205–4213.
- Sissi, C.; Palumbo, M. Effects of magnesium and related divalent metal ions in topoisomerase structure and function. Nucleic Acids Res. 2009, 37, 702–711.
- Deweese, J.E.; Burgin, A.B.; Osheroff, N. Human topoisomerase IIα uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res. 2008, 36, 4883–4893.
- Pitts, S.L.; Liou, G.F.; Mitchenall, L.A.; Burgin, A.B.; Maxwell, A.; Neuman, K.C.; Osheroff, N. Use of divalent metal ions in the DNA cleavage reaction of topoisomerase IV. Nucleic Acids Res. 2011, 39, 4808–4817.
- Dong, K.C.; Berger, J.M. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature 2007, 450, 1201–1205.
- Jang, Y.; Son, H.; Lee, S.W.; Hwang, W.; Jung, S.R.; Byl, J.A.W.; Osheroff, N.; Lee, S. Selection of DNA cleavage sites by topoisomerase II results from enzyme-induced flexibility of DNA. Cell. Chem. Biol. 2019, 26, 502–511.e3.
- Lee, S.; Jung, S.R.; Heo, K.; Byl, J.A.; Deweese, J.E.; Osheroff, N.; Hohng, S. DNA cleavage and opening reactions of human topoisomerase IIα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2925–2930.
- Lee, I.; Dong, K.C.; Berger, J.M. The role of DNA bending in type IIA topoisomerase function. Nucleic Acids Res. 2013, 41, 5444–5456.
- Corbett, K.D.; Shultzaberger, R.K.; Berger, J.M. The C-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl. Acad. Sci. USA 2004, 101, 7293–7298.
- Kramlinger, V.M.; Hiasa, H. The “GyrA-box” is required for the ability of DNA gyrase to wrap DNA and catalyze the supercoiling reaction. J. Biol. Chem. 2006, 281, 3738–3742.
- Lanz, M.A.; Klostermeier, D. The GyrA-box determines the geometry of DNA bound to gyrase and couples DNA binding to the nucleotide cycle. Nucleic Acids Res. 2012, 40, 10893–10903.
- Gibson, E.G.; Ashley, R.E.; Kerns, R.J.; Osheroff, N. Fluoroquinolone interactions with bacterial type II topoisomerases and target-mediated drug resistance. In Antimicrobial Resistance and Implications for the 21st Century; Drlica, K., Shlaes, D., Fong, I.W., Eds.; Springer: New York, NY, USA, 2018; pp. 507–529.
- Vos, S.M.; Lee, I.; Berger, J.M. Distinct regions of the Escherichia coli ParC C-terminal domain are required for substrate discrimination by topoisomerase IV. J. Mol. Biol. 2013, 425, 3029–3045.
- Tretter, E.M.; Lerman, J.C.; Berger, J.M. A naturally chimeric type IIA topoisomerase in Aquifex aeolicus highlights an evolutionary path for the emergence of functional paralogs. Proc. Natl. Acad. Sci. USA 2010, 107, 22055–22059.
- Corbett, K.D.; Schoeffler, A.J.; Thomsen, N.D.; Berger, J.M. The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 2005, 351, 545–561.
- Linka, R.M.; Porter, A.C.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-terminal regions of topoisomerase IIα and IIβ determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822.
- Broeck, A.V.; Lotz, C.; Drillien, R.; Haas, L.; Bedez, C.; Lamour, V. Structural basis for allosteric regulation of human topoisomerase IIα. Nat. Commun. 2021, 12, 2962.
- Antoniou-Kourounioti, M.; Mimmack, M.L.; Porter, A.C.G.; Farr, C.J. The impact of the C-terminal region on the interaction of topoisomerase IIα with mitotic chromatin. Int. J. Mol. Sci. 2019, 20, 1238.
- Deweese, J.E.; Osheroff, N. The use of divalent metal ions by type II topoisomerases. Metallomics 2010, 2, 450–459.
- Deweese, J.E.; Guengerich, F.P.; Burgin, A.B.; Osheroff, N. Metal ion interactions in the DNA cleavage/ligation active site of human topoisomerase IIα. Biochemistry 2009, 48, 8940–8947.
- Lindsley, J.E.; Wang, J.C. On the coupling between ATP usage and DNA transport by yeast DNA topoisomerase II. J. Biol. Chem. 1993, 268, 8096–8104.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
17 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No