 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pasquale Pagliaro | -- | 1883 | 2023-07-13 07:29:49 | | | |
| 2 | Conner Chen | Meta information modification | 1883 | 2023-07-17 04:22:17 | | |
Video Upload Options
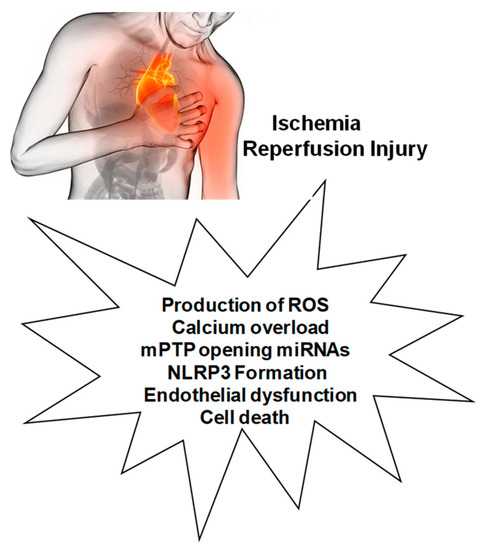
Myocardial ischemia-reperfusion injury (MIRI) is caused by several mechanisms, including the production of reactive oxygen species (ROS), altered cellular osmolarity, and inflammatory response. Calcium overload, altered oxygen levels, and mitochondrial ROS are also involved in these MIRI processes, resulting in the irreversible opening of the mitochondrial permeability transition pore (mPTP). These mechanisms and processes are associated with NLRP3 inflammasome priming and activation, which can also induce cell death by pyroptosis through the up-regulation of the caspase-1 pathway and IL-18 release. In addition, endothelial dysfunction, both in the presence and absence of MIRI, is also accompanied by altered oxygen levels, decreased nitric oxide production, and ROS overproduction, resulting in the expression of adhesion molecules and leukocyte infiltration in which the NLRP3 inflammasome plays a central role, thus contributing, through endothelial dysfunction, to the alteration of coronary flow, typical of ischemic heart disease. Given the intricate interrelationship between ROS and NLRP3, ROS inhibitors can reduce NLRP3 inflammasome activation, while NLRP3 inhibitors can reduce oxidative stress and inflammation.
1. Introduction
MIRI Activates Various Cell Death Pathways
2. Mechanisms Involved in MIRI and Interactions with NLRP3
2.1. Production of ROS
2.2. Calcium Overload
2.3. Role of mPTP Opening
2.4. Endothelial Dysfunction
References
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266.
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99.
- Penna, C.; Comità, S.; Tullio, F.; Alloatti, G.; Pagliaro, P. Challenges facing the clinical translation of cardioprotection: 35 years after the discovery of ischemic preconditioning. Vascul. Pharmacol. 2022, 144, 106995.
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142.
- Pasqua, T.; Pagliaro, P.; Rocca, C.; Angelone, T.; Penna, C. Role of NLRP-3 Inflammasome in Hypertension: A Potential Therapeutic Target. Curr. Pharm. Biotechnol. 2018, 19, 708–714.
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401.
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.O.; Garcia-Dorado, D.; EU-CARDIOPROTECTION COST Action (CA16225). Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795–3806.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Mastrocola, R.; Aragno, M.; Alloatti, G.; Collino, M.; Penna, C.; Pagliaro, P. Metaflammation: Tissue-Specific Alterations of the NLRP3 Inflammasome Platform in Metabolic Syndrome. Curr. Med. Chem. 2018, 25, 1294–1310.
- Zhao, J.; Li, J.; Li, G.; Chen, M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 9, 1059576.
- Liu, Y.; Zhang, J.; Zhang, D.; Yu, P.; Zhang, J.; Yu, S. Research Progress on the Role of Pyroptosis in Myocardial Ischemia-Reperfusion Injury. Cells 2022, 11, 3271.
- Popov, S.V.; Maslov, L.N.; Naryzhnaya, N.V.; Mukhomezyanov, A.V.; Krylatov, A.V.; Tsibulnikov, S.Y.; Ryabov, V.V.; Cohen, M.V.; Downey, J.M. The Role of Pyroptosis in Ischemic and Reperfusion Injury of the Heart. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 562–574.
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568.
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5.
- Penna, C.; Perrelli, M.G.; Pagliaro, P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: Therapeutic implications. Antioxid. Redox Signal. 2013, 18, 556–599.
- Liu, C.; Li, Z.; Li, B.; Liu, W.; Zhang, S.; Qiu, K.; Zhu, W. Relationship be-tween ferroptosis and mitophagy in cardiac ischemia reperfusion injury: A mini-review. PeerJ 2023, 11, e14952.
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569.
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043.
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden”. Cell Death Cells 2020, 9, 1505.
- Pagliaro, P.; Moro, F.; Tullio, F.; Perrelli, M.G.; Penna, C. Cardioprotective pathways during reperfusion: Focus on redox signaling and other modalities of cell signaling. Antioxid. Redox Signal. 2011, 14, 833–850.
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell. Longev. 2016, 2016, 2183026.
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol. 2006, 101, 180–189.
- Downey, J.M.; Cohen, M.V. A really radical observation: A comment on Penna et al. in Basic Res Cardiol (2006) 101:180-189. Basic Res. Cardiol. 2006, 101, 190–191.
- Tsutsumi, Y.M.; Yokoyama, T.; Horikawa, Y.; Roth, D.M.; Patel, H.H. Reactive oxygen species trigger ischemic and pharmacological postconditioning: In vivo and in vitro characterization. Life Sci. 2007, 81, 1223–1227.
- Zuurbier, C.J. NLRP3 Inflammasome in Cardioprotective Signaling. J. Cardiovasc. Pharmacol. 2019, 74, 271–275.
- Zuurbier, C.J.; Jong, W.M.; Eerbeek, O.; Koeman, A.; Pulskens, W.P.; Butter, L.M.; Leemans, J.C.; Hollmann, M.W. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased IL-6/STAT3 signaling. PLoS ONE 2012, 7, e40643.
- Sandanger, Ø.; Gao, E.; Ranheim, T.; Bliksøen, M.; Kaasbøll, O.J.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.K.; Attramadal, H.; et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020.
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.H. The role of Ca2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Investig. 2019, 99, 499–513.
- Mo, G.; Liu, X.; Zhong, Y.; Mo, J.; Li, Z.; Li, D.; Zhang, L.; Liu, Y. IP3R1 regulates Ca2+ transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 31.
- García-Niño, W.R.; Zazueta, C.; Buelna-Chontal, M.; Silva-Palacios, A. Mitochondrial Quality Control in Cardiac-Conditioning Strategies against Ischemia-Reperfusion Injury. Life 2021, 11, 1123.
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093.
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat. Commun. 2019, 10, 4341.
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial permeability transition pore-dependent necrosis. J. Mol. Cell. Cardiol. 2023, 174, 47–55.
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789.
- Heusch, G. Coronary microvascular obstruction: The new frontier in cardioprotection. Basic Res. Cardiol. 2019, 114, 45.
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791.
- Huertas, J.; Lee, H.T. Multi-faceted roles of cathepsins in ischemia reperfusion injury (Review). Mol. Med. Rep. 2022, 26, 368.
- Sabri, A.; Alcott, S.G.; Elouardighi, H.; Pak, E.; Derian, C.; Andrade-Gordon, P.; Kinnally, K.; Steinberg, S.F. Neutrophil cathepsin G promotes detachment-induced cardiomyocyte apoptosis via a protease-activated receptor-independent mechanism. J. Biol. Chem. 2003, 278, 23944–23954.
- Hooshdaran, B.; Kolpakov, M.A.; Guo, X.; Miller, S.A.; Wang, T.; Tilley, D.G.; Rafiq, K.; Sabri, A. Dual inhibition of cathepsin G and chymase reduces myocyte death and improves cardiac remodeling after myocardial ischemia reperfusion injury. Basic Res. Cardiol. 2017, 112, 62.

