 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nidhi Gour | -- | 4130 | 2023-07-12 04:50:24 | | | |
| 2 | Lindsay Dong | Meta information modification | 4130 | 2023-07-13 03:01:54 | | |
Video Upload Options
Amyloids were conventionally referred to as extracellular and intracellular accumulation of Aβ42 peptide, which causes the formation of plaques and neurofibrillary tangles inside the brain leading to the pathogenesis in Alzheimer’s disease. Subsequently, amyloid-like deposition was found in the etiology of prion diseases, Parkinson’s disease, type II diabetes, and cancer, which was attributed to the aggregation of prion protein, α-Synuclein, islet amyloid polypeptide protein, and p53 protein, respectively. Hence, traditionally amyloids were considered aggregates formed exclusively by proteins or peptides. However, since the last decade, it has been discovered that other metabolites, like single amino acids, nucleobases, lipids, glucose derivatives, etc., have a propensity to form amyloid-like toxic assemblies. Several studies suggest direct implications of these metabolite assemblies in the patho-physiology of various inborn errors of metabolisms like phenylketonuria, tyrosinemia, cystinuria, and Gaucher’s disease, to name a few.
1. Introduction
2. General Characteristics of Amyloid Structures
3. Protein Aggregation in Alzheimer’s Disease (AD)
4. α-Synuclein Aggregation and Parkinson’s Disease
5. Aggregation of Prion Protein (PrP) and Associated Diseases
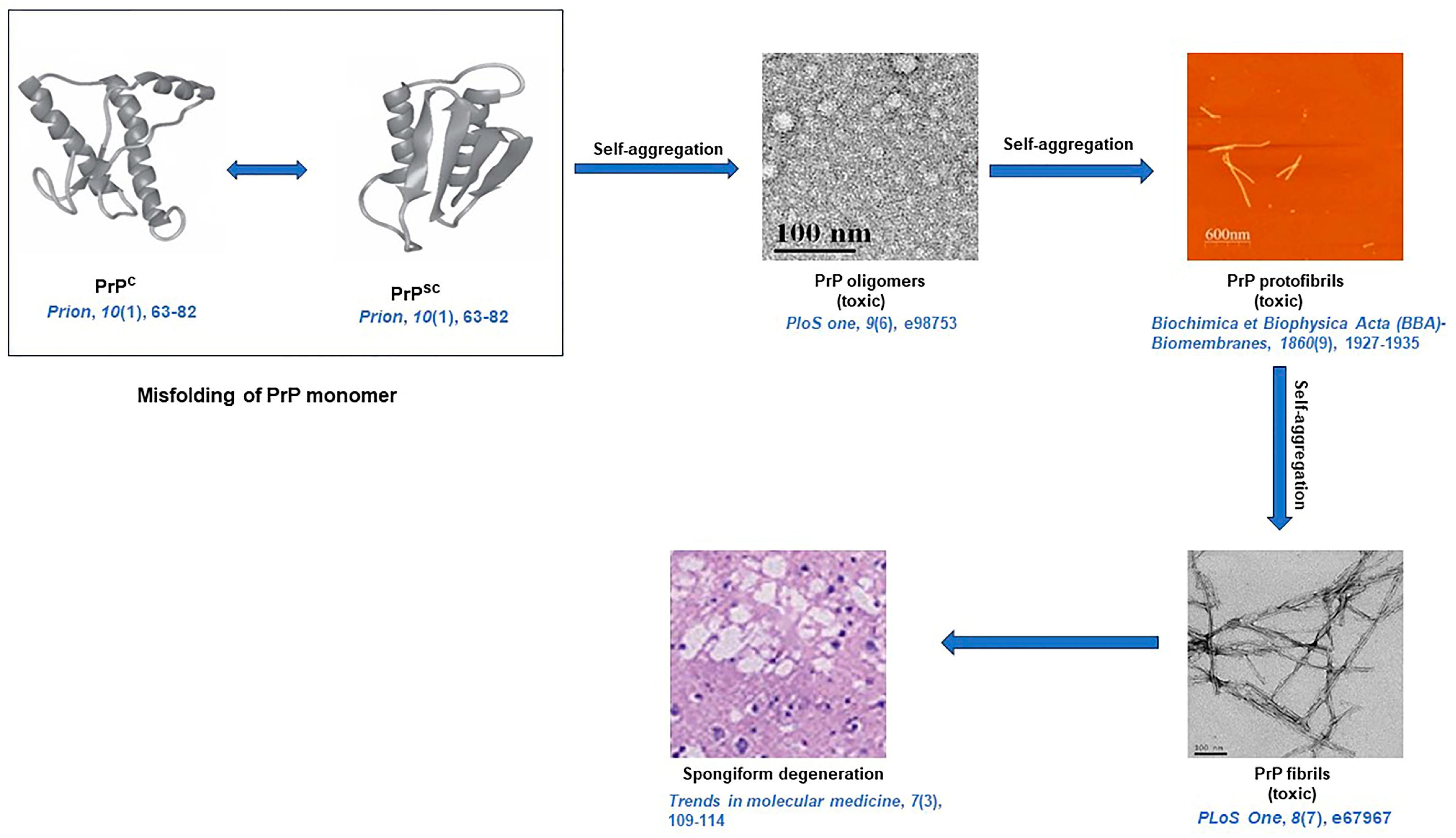
6. IAPP Aggregation and Type 2 Diabetes
7. Amyloid-like Aggregation in the Patho-Physiology of Cancer
8. Metabolites Assemblies as a Surprising Extension to Generic Amyloid Hypothesis
8.1. Amyloid-like Aggregation of Single Amino Acids and Its Implications in IEMs
8.2. Non-Proteinaceous Metabolite Assemblies and Their Implications in IEMs
9. Functional Amyloid
10. Amyloid Cross-Seeding and Interaction
11. Critical Analysis and Future Outlook
References
- Kyle, R.A. Amyloidosis: A convoluted story. Br. J. Haematol. 2001, 114, 529–538.
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773.
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431.
- Plascencia-Villa, G.; Perry, G. Neuropathological changes provide insights into key mechanisms related to Alzheimer’s disease and related dementia. Am. J. Pathol. 2022, 192, 1340–1346.
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946.
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13.
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M. Islet amyloid polypeptide: Structure, function, and pathophysiology. J. Diabetes Res. 2016, 2016, 2798269.
- Masliah, E.; Rockenstein, E.; Inglis, C.; Adame, A.; Bett, C.; Lucero, M.; Sigurdson, C.J. Prion infection promotes extensive accumulation of α-synuclein in aged human α-synuclein transgenic mice. Prion 2012, 6, 184–190.
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132.
- Wille, H.; Requena, J.R. The structure of PrPSc prions. Pathogens 2018, 7, 20.
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84.
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer disease amyloid-β peptide and other amyloid proteins: A further aspect of the amyloid cascade hypothesis. J. Biol. Chem. 2016, 291, 16485–16493.
- Konstantoulea, K.; Louros, N.; Rousseau, F.; Schymkowitz, J. Heterotypic interactions in amyloid function and disease. FEBS J. 2022, 289, 2025–2046.
- Cherny, I.; Gazit, E. Amyloids: Not only pathological agents but also ordered nanomaterials. Angew. Chem. Int. Ed. 2008, 47, 4062–4069.
- Levkovich, S.A.; Gazit, E.; Bar-Yosef, D.L. Two decades of studying functional amyloids in microorganisms. Trends Microbiol. 2021, 29, 251–265.
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706.
- Gour, N.; Kanth, P.C.; Koshti, B.; Kshtriya, V.; Shah, D.; Patel, S.; Agrawal-Rajput, R.; Pandey, M.K. Amyloid-like structures formed by single amino acid self-assemblies of cysteine and methionine. ACS Chem. Neurosci. 2018, 10, 1230–1239.
- Koshti, B.; Kshtriya, V.; Singh, R.; Walia, S.; Bhatia, D.; Joshi, K.B.; Gour, N. Unusual Aggregates Formed by the Self-Assembly of Proline, Hydroxyproline, and Lysine. ACS Chem. Neurosci. 2021, 12, 3237–3249.
- Shaham-Niv, S.; Adler-Abramovich, L.; Schnaider, L.; Gazit, E. Extension of the generic amyloid hypothesis to nonproteinaceous metabolite assemblies. Sci. Adv. 2015, 1, e1500137.
- Koshti, B.; Kshtriya, V.; Naskar, S.; Narode, H.; Gour, N. Controlled aggregation properties of single amino acids modified with protecting groups. New J. Chem. 2022, 46, 4746–4755.
- Gour, N.; Gazit, E. Metabolite assemblies: A surprising extension to the amyloid hypothesis. Curr. Opin. Chem. Biol. 2021, 64, 154–164.
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235.
- Khurana, R.; Coleman, C.; Ionescu-Zanetti, C.; Carter, S.A.; Krishna, V.; Grover, R.K.; Roy, R.; Singh, S. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 2005, 151, 229–238.
- Kashyap, P.; Kalaiselvan, V.; Kumar, R.; Kumar, S. Ajmalicine and reserpine: Indole alkaloids as multi-target directed ligands towards factors implicated in Alzheimer’s disease. Molecules 2020, 25, 1609.
- Amdursky, N.; Stevens, M.M. Circular dichroism of amino acids: Following the structural formation of phenylalanine. ChemPhysChem 2015, 16, 2768–2774.
- Carulla, N.; Zhou, M.; Arimon, M.; Gairí, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Experimental characterization of disordered and ordered aggregates populated during the process of amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2009, 106, 7828–7833.
- Adamcik, J.; Mezzenga, R. Study of amyloid fibrils via atomic force microscopy. Curr. Opin. Colloid Interface Sci. 2012, 17, 369–376.
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699.
- Arispe, N.; Pollard, H.B.; Rojas, E. The Ability of Amyloid β-Protein to Form Ca2+ Channels Provides a Mechanism for Neuronal Death in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1994, 747, 256–266.
- Wells, C.; Brennan, S.; Keon, M.; Ooi, L. The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 2021, 181, 582–604.
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666.
- Ghiso, J.; Frangione, B. Amyloidosis and Alzheimer’s disease. Adv. Drug Deliv. Rev. 2002, 54, 1539–1551.
- Liang, Y.; Wang, W.; Sun, Y.; Dong, X. Insights into the cross-amyloid aggregation of Aβ40 and its N-terminal truncated peptide Aβ11–40 affected by epigallocatechin gallate. Chin. J. Chem. Eng. 2022, 45, 284–293.
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409.
- Sultan, F.; Parkin, E.T. The amyloid precursor protein plays differential roles in the UVA resistance and proliferation of human retinal pigment epithelial cells. Protein Pept. Lett. 2022, 29, 313–327.
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid β protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282.
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193.
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2000, 1502, 16–30.
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T., Jr. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125.
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760.
- Morris, K.L.; Serpell, L.C. X-ray fibre diffraction studies of amyloid fibrils. Amyloid Proteins Methods Protoc. 2012, 849, 121–135.
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674.
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Jiang, Z.; Huang, Y.; Zhang, P.; Han, C.; Lu, Y.; Mo, Z.; Zhang, Z.; Li, X.; Zhao, S.; Cai, F. Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 2020, 13, 102.
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644.
- Zhang, S.; Liu, Y.-Q.; Jia, C.; Lim, Y.-J.; Feng, G.; Xu, E.; Long, H.; Kimura, Y.; Tao, Y.; Zhao, C. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2011196118.
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254.
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715.
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449.
- Meena, V.K.; Kumar, V.; Karalia, S.; Dangi, R.S.; Sundd, M. Structural and mechanistic insights into modulation of α-Synuclein fibril formation by aloin and emodin. Biochim. Biophys. Acta (BBA) Gen. Subj. 2022, 1866, 130151.
- George, J.M. The synucleins. Genome Biol. 2001, 3, 1–6.
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48.
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415.
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402.
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Batlle, C.; Ventura, S. Amyloids or prions? That is the question. Prion 2015, 9, 200–206.
- Kushnirov, V.V.; Vishnevskaya, A.B.; Alexandrov, I.M.; Ter-Avanesyan, M.D. Prion and nonprion amyloids: A comparison inspired by the yeast Sup35 protein. Prion 2007, 1, 179–184.
- Ghetti, B.; Piccardo, P.; Frangione, B.; Bugiani, O.; Giaccone, G.; Young, K.; Prelli, F.; Farlow, M.R.; Dlouhy, S.R.; Tagliavini, F. Prion protein amyloidosis. Brain Pathol. 1996, 6, 127–145.
- Stöhr, J.; Weinmann, N.; Wille, H.; Kaimann, T.; Nagel-Steger, L.; Birkmann, E.; Panza, G.; Prusiner, S.B.; Eigen, M.; Riesner, D. Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. USA 2008, 105, 2409–2414.
- Chatterjee, B.; Lee, C.-Y.; Lin, C.; Chen, E.H.-L.; Huang, C.-L.; Yang, C.-C.; Chen, R.P.-Y. Amyloid core formed of full-length recombinant mouse prion protein involves sequence 127–143 but not sequence 107–126. PLoS ONE 2013, 8, e67967.
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235.
- Yamaguchi, K.-I.; Kuwata, K. Formation and properties of amyloid fibrils of prion protein. Biophys. Rev. 2018, 10, 517–525.
- Atkinson, C.J.; Zhang, K.; Munn, A.L.; Wiegmans, A.; Wei, M.Q. Prion protein scrapie and the normal cellular prion protein. Prion 2016, 10, 63–82.
- Ladner-Keay, C.L.; Griffith, B.J.; Wishart, D.S. Shaking alone induces de novo conversion of recombinant prion proteins to β-sheet rich oligomers and fibrils. PLoS ONE 2014, 9, e98753.
- Thody, S.A.; Mathew, M.K.; Udgaonkar, J.B. Mechanism of aggregation and membrane interactions of mammalian prion protein. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 1927–1935.
- Soto, C.; Saborio, G.P. Prions: Disease propagation and disease therapy by conformational transmission. Trends Mol. Med. 2001, 7, 109–114.
- Ferreira, S.; Raimundo, A.F.; Menezes, R.; Martins, I.C. Islet amyloid polypeptide & amyloid beta peptide roles in Alzheimer’s disease: Two triggers, one disease. Neural Regen. Res. 2021, 16, 1127.
- Novials, A.; Rojas, I.; Casamitjana, R.; Usac, E.; Gomis, R. A novel mutation in islet amyloid polypeptide (IAPP) gene promoter is associated with Type II diabetes mellitus. Diabetologia 2001, 44, 1064–1065.
- O’Brien, T.; Butler, P.; Westermark, P.; Johnson, K. Islet amyloid polypeptide: A review of its biology and potential roles in the pathogenesis of diabetes mellitus. Vet. Pathol. 1993, 30, 317–332.
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760.
- Howard, C.F., Jr. Insular amyloidosis and diabetes mellitus in Macaca nigra. Diabetes 1978, 27, 357–364.
- Betsholtz, C.; Christmansson, L.; Engström, U.; Rorsman, F.; Svensson, V.; Johnson, K.H.; Westermark, P. Sequence divergence in a specific region of islet amyloid polypeptide (IAPP) explains differences in islet amyloid formation between species. FEBS Lett. 1989, 251, 261–264.
- Westermark, P. Quantitative studies of amyloid in the islets of Langerhans. Upsala J. Med. Sci. 1972, 77, 91–94.
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. FEBS J. 2005, 272, 5950–5961.
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and drug targeting insights on mutant p53. Cancers 2021, 13, 3344.
- Billant, O.; Friocourt, G.; Roux, P.; Voisset, C. p53, A Victim of the Prion Fashion. Cancers 2021, 13, 269.
- Bom, A.P.A.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162.
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798.
- de Oliveira, G.A.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.; Cino, E.A.; Silva, J.L. The status of p53 oligomeric and aggregation states in cancer. Biomolecules 2020, 10, 548.
- Navalkar, A.; Pandey, S.; Singh, N.; Patel, K.; Datta, D.; Mohanty, B.; Jadhav, S.; Chaudhari, P.; Maji, S.K. Direct evidence of cellular transformation by prion-like p53 amyloid infection. J. Cell Sci. 2021, 134, jcs258316.
- Aliu, E.; Kanungo, S.; Arnold, G.L. Amino acid disorders. Ann. Transl. Med. 2018, 6, 471.
- Ménard-Moyon, C.; Venkatesh, V.; Krishna, K.V.; Bonachera, F.; Verma, S.; Bianco, A. Self-Assembly of Tyrosine into Controlled Supramolecular Nanostructures. Chem. Eur. J. 2015, 21, 11681–11686.
- Sade Yazdi, D.; Laor Bar-Yosef, D.; Adsi, H.; Kreiser, T.; Sigal, S.; Bera, S.; Zaguri, D.; Shaham-Niv, S.; Oluwatoba, D.S.; Levy, D. Homocysteine fibrillar assemblies display cross-talk with Alzheimer’s disease β-amyloid polypeptide. Proc. Natl. Acad. Sci. USA 2021, 118, e2017575118.
- Zaguri, D.; Shaham-Niv, S.; Naaman, E.; Mimouni, M.; Magen, D.; Pollack, S.; Kreiser, T.; Leibu, R.; Rencus-Lazar, S.; Adler-Abramovich, L. Induction of retinopathy by fibrillar oxalate assemblies. Commun. Chem. 2020, 3, 2.
- Paul, A.; Jacoby, G.; Laor Bar-Yosef, D.; Beck, R.; Gazit, E.; Segal, D. Glucosylceramide Associated with Gaucher Disease Forms Amyloid-like Twisted Ribbon Fibrils That Induce α-Synuclein Aggregation. ACS Nano 2021, 15, 11854–11868.
- Anand, B.G.; Prajapati, K.P.; Dubey, K.; Ahamad, N.; Shekhawat, D.S.; Rath, P.C.; Joseph, G.K.; Kar, K. Self-assembly of artificial sweetener aspartame yields amyloid-like cytotoxic nanostructures. ACS Nano 2019, 13, 6033–6049.
- DeBenedictis, E.P.; Liu, J.; Keten, S. Adhesion mechanisms of curli subunit CsgA to abiotic surfaces. Sci. Adv. 2016, 2, e1600998.
- Edwards, S.J.; Kjellerup, B.V. Applications of biofilms in bioremediation and biotransformation of persistent organic pollutants, pharmaceuticals/personal care products, and heavy metals. Appl. Microbiol. Biotechnol. 2013, 97, 9909–9921.
- Donato, V.; Ayala, F.R.; Cogliati, S.; Bauman, C.; Costa, J.G.; Lenini, C.; Grau, R. Bacillus subtilis biofilm extends Caenorhabditis elegans longevity through downregulation of the insulin-like signalling pathway. Nat. Commun. 2017, 8, 14332.
- Hobley, L.; Harkins, C.; MacPhee, C.E.; Stanley-Wall, N.R. Giving structure to the biofilm matrix: An overview of individual strategies and emerging common themes. FEMS Microbiol. Rev. 2015, 39, 649–669.
- Yates, M.D.; Strycharz-Glaven, S.M.; Golden, J.P.; Roy, J.; Tsoi, S.; Erickson, J.S.; El-Naggar, M.Y.; Barton, S.C.; Tender, L.M. Measuring conductivity of living Geobacter sulfurreducens biofilms. Nat. Nanotechnol. 2016, 11, 910–913.
- Shimanovich, U.; Michaels, T.C.; De Genst, E.; Matak-Vinkovic, D.; Dobson, C.M.; Knowles, T.P. Sequential release of proteins from structured multishell microcapsules. Biomacromolecules 2017, 18, 3052–3059.
- Smith, J.F.; Knowles, T.P.; Dobson, C.M.; MacPhee, C.E.; Welland, M.E. Characterization of the nanoscale properties of individual amyloid fibrils. Proc. Natl. Acad. Sci. USA 2006, 103, 15806–15811.
- Kol, N.; Adler-Abramovich, L.; Barlam, D.; Shneck, R.Z.; Gazit, E.; Rousso, I. Self-assembled peptide nanotubes are uniquely rigid bioinspired supramolecular structures. Nano Lett. 2005, 5, 1343–1346.
- Knowles, T.P.; Buehler, M.J. Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 2011, 6, 469–479.
- Keten, S.; Xu, Z.; Ihle, B.; Buehler, M.J. Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk. Nat. Mater. 2010, 9, 359–367.
- Zhao, J.; Yang, P. Amyloid-Mediated Fabrication of Organic–Inorganic Hybrid Materials and Their Biomedical Applications. Adv. Mater. Interfaces 2020, 7, 2001060.
- Morales, R.; Green, K.M.; Soto, C. Cross currents in protein misfolding disorders: Interactions and therapy. CNS Neurol. Disord. Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2009, 8, 363–371.
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636.
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118.
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74.
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.-X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64, 570–575.
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid cross-seeding: Mechanism, implication, and inhibition. Molecules 2022, 27, 1776.
- Gabr, M.T.; Peccati, F. Dual targeting of monomeric tau and α-synuclein aggregation: A new multitarget therapeutic strategy for neurodegeneration. ACS Chem. Neurosci. 2020, 11, 2051–2057.
- Di Giovanni, S.; Eleuteri, S.; Paleologou, K.E.; Yin, G.; Zweckstetter, M.; Carrupt, P.-A.; Lashuel, H.A. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of α-synuclein and β-amyloid and protect against amyloid-induced toxicity. J. Biol. Chem. 2010, 285, 14941–14954.
- Ono, K.; Yamada, M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. J. Neurochem. 2006, 97, 105–115.
- Bruno, E.; Pereira, C.; Roman, K.P.; Takiguchi, M.; Kao, P.-Y.; Nogaj, L.A.; Moffet, D.A. IAPP aggregation and cellular toxicity are inhibited by 1,2,3,4,6-penta-O-galloyl-β-ᴅ-glucose. Amyloid 2013, 20, 34–38.
- de Almeida, N.E.; Do, T.D.; LaPointe, N.E.; Tro, M.; Feinstein, S.C.; Shea, J.-E.; Bowers, M.T. 1,2,3,4,6-penta-O-galloyl-β-ᴅ-glucopyranose binds to the N-terminal metal binding region to inhibit amyloid β-protein oligomer and fibril formation. Int. J. Mass Spectrom. 2017, 420, 24–34.

