Amyloids were conventionally referred to as extracellular and intracellular accumulation of Aβ42 peptide, which causes the formation of plaques and neurofibrillary tangles inside the brain leading to the pathogenesis in Alzheimer’s disease. Subsequently, amyloid-like deposition was found in the etiology of prion diseases, Parkinson’s disease, type II diabetes, and cancer, which was attributed to the aggregation of prion protein, α-Synuclein, islet amyloid polypeptide protein, and p53 protein, respectively. Hence, traditionally amyloids were considered aggregates formed exclusively by proteins or peptides. However, since the last decade, it has been discovered that other metabolites, like single amino acids, nucleobases, lipids, glucose derivatives, etc., have a propensity to form amyloid-like toxic assemblies. Several studies suggest direct implications of these metabolite assemblies in the patho-physiology of various inborn errors of metabolisms like phenylketonuria, tyrosinemia, cystinuria, and Gaucher’s disease, to name a few.
1. Introduction
In 1838, German botanist Matthias Schleiden, for the very first time, introduced the term “amyloid” to the scientific literature to represent the amylaceous constituent of plants [
1]. Later in 1854, Rudolph Virchow coined the term “amyloid” to describe the tissue abnormality that displayed an iodine-staining reaction [
2]. Further, in 1907, German psychiatrist Aloysius (Alois) Alzheimer referred to plaques and neurofibrillary tangles present in the brain of patients with senile dementia as “amyloid” [
3,
4]. These abnormal aggregates were formed by the aggregation of Aβ42 and Aβ40 peptides, and the disease was later referred to as Alzheimer’s disease (AD) [
5]. Subsequently, aggregation of tau oligomers was also considered a hallmark in the progression of AD [
6]. Similar protein aggregation was also discovered in Parkinson’s disease (PD), wherein α-synuclein (α-Syn) protein aggregates inside the brain leading to pathogenesis like dementia and memory loss [
7]. Further, it was also discovered that islet amyloid polypeptide (IAPP) aggregates to fibrillar structures leading to type II diabetes [
8]. Research on studying the progression of the plethora of prion diseases suggests the formation of the infectious prion protein (PrP) with a tendency to cross-seed aggregation in other proteins [
9,
10]. Bovine spongiform encephalopathy (BSE or “mad cow” disease) in cattle, Creutzfeldt–Jakob disease (CJD) and variant CJD in humans, scrapie in sheep, and chronic wasting disease (CWD) in deer, elk, mice, and reindeer are some of the diseases associated with PrP aggregation [
11]. A similar aggregation observed for the PrP protein was also noted for the p53 protein in the patho-physiology of cancer [
12].
Several cross interaction studies of these amyloids have also been pursued [
13,
14] and functional materials have been designed using amyloid fibers due to their exceptional mechanical strength [
15,
16].
Initially, it was thought that amyloids are formed exclusively by the aggregation of proteins and peptides. However, several literature reports in the last decade implicate that single amino acids and non-proteinaceous metabolites also exhibit a tendency to aggregate and form amyloid-like structures [
17,
18,
19,
20,
21]. Interestingly, the accumulation of metabolites reveals a similar aggregation pathway as that exhibited by conventional amyloidogenic proteins and peptides such as Aβ42, PrP, and α-Syn [
22]. Hence, it may be surmised that the patho-physiology of rare inborn errors of metabolisms (IEMs) may have a common etiology for amyloid-associated diseases [
22]. The research group of Gazit and coworkers has worked extensively on metabolite assemblies research and reported amyloid-like toxic aggregates formed by single amino acids like phenylalanine, tyrosine, tryptophan, and non-proteinaceous metabolites like uracil, orotic acid, adenine, and oxalic acid, and proposed a “Generic amyloid Hypothesis” which implicates amyloid-like structure formation as a general phenomenon in the patho-physiology of diseases [
17,
20].
2. General Characteristics of Amyloid Structures
The aggregates which are formed by amyloid have some common characteristics like binding to amyloid-specific dyes Congo red (CR) and Thioflavin T (ThT) [
23,
24]. Typically, amyloidogenic peptides or proteins are rich in β-sheets and hence produce a characteristic negative band around 220 nm in circular dichroism (CD) [
25,
26]. The morphology of amyloid-like aggregates has been studied by microscopic techniques like transmission electron microscopy (TEM), scanning electron microscopy (SEM), atomic force microscopy (AFM), and light microscopy [
18,
19,
20,
21,
27,
28]. amyloid-like aggregation is commonly associated with the formation of varying morphologies (polymorphs) during the course of disease progression [
19]. The early aggregates are usually spherulite-like structures that gradually transform into fibrillar assemblies. The early aggregates, i.e., the pre-fibrillar aggregates of Aβ-peptide, huntingtin, α-synuclein, and transthyretin, are the most toxic and infectious ones [
20,
29]. These early aggregates impair cellular functions by interacting with cell membranes and causing oxidative stress. Furthermore, pre-fibrillar aggregates increase free Ca
2+ ion concentration, which eventually leads to apoptotic or neural cell death [
30]. Several studies also suggest that not only the pre-fibrillar aggregates but the soluble oligomers of many proteins or peptides are also toxic in major amyloid diseases, for instance, spongiform encephalopathies, Huntington disease, type II diabetes, AD, and PD [
31].
3. Protein Aggregation in Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is associated with the formation of senile plaques and neurofibrillary tangles in the brain, which causes dementia and memory loss [
4]. The plaques are the extracellular deposits of amyloid β (Aβ) protein, while the neurofibrillary tangles are intracellular accumulations [
32]. Aβ42 is a peptide formed by 42 amino acids and is the main constituent of the lesions found in the brain of patients with AD, whereas Aβ40 is the most abundant isoform constituted by 40 amino acid sequences [
33,
34]. Astrocytes and neurons produce Aβ peptides in the brain by the proteolytic processing of the β-amyloid precursor protein (APP) mediated by enzymes such as β-secretase and γ-secretase [
35]. It is well-documented that the astrocytes affected by AD express high levels of APP, β-secretase, and γ-secretase, the three main components required for amyloid production [
29]. Proteolysis of APP by α-secretase and β-secretase leads to the secretion of sAPPα and sAPPβ, respectively [
36]. The secreted sAPPα or sAPPβ have C-terminal fragments which can be cleaved by γ-secretase extracellularly to release the peptides Aβ42 and Aβ40 [
36].
Yankner et al. investigated the neurotoxicity produced by Aβ fibrils on hippocampal neuronal cell lines by co-incubation studies under different time periods. Their study suggests that both concentration and period of co-incubation of Aβ40 fibrils with the neuronal cells play a crucial role in predicting the trophic and toxic response of Aβ40 and its effect on neuronal differentiation [
41]. In a recent study, Antonino et al. reported the amyloidogenic processing of APP by the oligomers and fibrils of Aβ. It was noted that exacerbated and intracellular accumulation of Aβ42 is caused due to colocalization and physical interaction of APP and BACE1. It was also noted that cells overexpressing the mutant forms of APP, which cannot bind to Aβ, could not increase the colocalization of APP with BACE1, indicating a crucial role of physical binding of Aβ to APP/BACE1 in causing amyloidogenic deposits.
In 1963, Kidd first reported the structure of amyloid fibrils present in the cerebral cortex of AD patients with the help of electron microscopy (EM). The micrograph revealed that amyloid fibrils exist as paired helical filaments coiled in a squash racket shape [
44]. The EM of Aβ42 and Aβ40 mature fibers suggest long straight fibrillar morphology having a diameter in the range of 70–80 Å [
45]. The negative-stain EM of protofibrils of Aβ42 and Aβ40 revealed flexible fibers having a diameter in the range of 60–100 Å. The structures of the Aβ42 and Aβ40 protofibrils were also analyzed by AFM, which revealed the diameter of Aβ40 protofibrils to be around 3.1 ± 0.9 nm and the diameter of Aβ40 long fibrillar species as 7.8 ± 0.45 nm [
46]. The diameter of Aβ42 protofibrils was found to be 4.2 ± 0.58 nm, while the diameter of bifurcated type-1 and type-2 fibrils of Aβ42 was found to be 7.3 ± 0.53 nm and 3.8 ± 0.43 nm, respectively. Kollmer et al. further reported the cryogenic electronic microscopy (Cryo-EM) structure of Aβ amyloid fibrils from meningeal Alzheimer’s brain tissue. Their studies suggest that Aβ amyloid fibrils have a polymorphic nature, and their morphology is right-hand twisted [
47]. The X-ray diffraction (XRD) analysis of amyloid fibers reveals a strong 4.8 Å reflection on the meridian, which corresponds to the hydrogen bonding distance between β-strands. The 10–11 Å reflection on the equator, on the other hand, corresponded to the inter-sheet distance of 10.7 Å [
48]. Griffin and his team reported the atomic resolution structure of monomorphic Aβ42 amyloid fibrils. The structure shows that the core of fibril consists of a dimer of Aβ42 molecules, each containing four β-strands in an S-shaped amyloid fold and arranged in a manner that generates two hydrophobic cores that are capped at the end of the chain by a salt bridge [
49]. Riek and coworkers presented the 3D structure of Aβ42 fibril, which revealed that residues of 15–42 in Aβ42 form a double horseshoe-like cross β-sheet structure [
50]. Circular dichroism (CD) analysis of aqueous solutions of Aβ42 and Aβ40 suggests the aggregates have mostly β-sheet structure [
45].
4. α-Synuclein Aggregation and Parkinson’s Disease
Parkinson’s disease (PD) was reported for the very first time by James Parkinson in 1817. The main pathological characteristics of PD are the formation of Lewy bodies inside the brain [
63]. The key components present in the Lewy bodies are misfolded α-Syn peptides [
64]. Jiang et al., through their studies, reported mutation in the synuclein alpha (
SNCA) gene as the main reason behind the abnormal aggregation of α-Syn in PD [
65]. The amyloidosis process of the α-syn peptide includes the formation of soluble oligomers, which further convert into insoluble amyloid fibrillar morphology by self-association [
66]. These fibrils are the pathological emblem of PD, and other synucleinopathies are formed due to protein misfolding and aggregation of α-Syn, a 140 amino acid long protein that is normally present at high levels in the brain and is embroiled in crucial synaptic processes in the neurons [
67,
68]. It is basically a disorganized protein but adopts a partial α-helix motif upon binding with membranes [
69]. The presence of the amphipathic region enables the α-Syn protein to bind with membranes [
70]. The amphipathic region of α-Syn is followed by the hydrophobic or non-amyloid component (NAC) region, which is mainly responsible for inducing its abnormal aggregation [
71]. It was found that β and γ-synuclein are the two isoforms of α-Syn [
72]. Dysregulation of cellular processes and toxic gain or losses of function are amongst the major molecular mechanisms that may account for the neurotoxicity associated with α-Syn aggregation [
73].
Tuttle et al. characterized α-Syn fibrils by using TEM analysis, which revealed that α-Syn fibrils have a width of 4.6 ± 0.4 nm. The X-ray diffraction analysis showed an archetypal meridional diffraction pattern at 4.8 Å, which corresponds to a cross β-sheet structure of α-Syn fibrils. The high-resolution 3D structure of a single untwisted α-Syn amyloid fibril in the substantia nigra of the brains of people who have PD reveals a diameter of around 5 nm as assessed through EM. The structure of a pathogenic fibril of full-length human α-Syn was studied through solid-state NMR and was validated by EM and XRD. These studies suggest that α-Syn fibrils exhibit typical amyloid features, which include parallel, in-register β-sheets and hydrophobic-core residues with substantial complexity arising from diverse structural features like an intermolecular salt bridge, a glutamine ladder, close backbone interactions involving small residues, and several steric zippers stabilizing a new orthogonal Greek-key topology. These characteristics contribute to the robust propagation of this fibril form, as supported by the structural similarity of early-onset PD mutants [
82]. Stahlberg and coworkers also elucidated the Cryo-EM structure of α-Syn amyloid fibrils (residues 1–121) at a resolution of 3.4 Å which reveals two protofilaments are intertwined in left-handed helix, and the protofilaments offer Greek-like topology [
83].
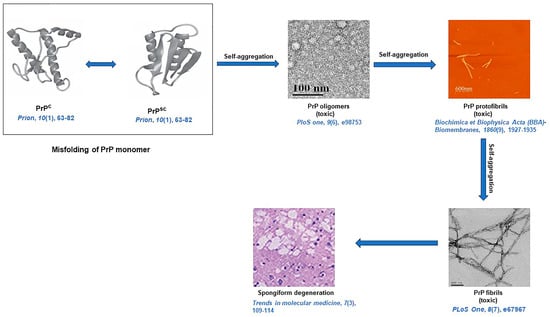
5. Aggregation of Prion Protein (PrP) and Associated Diseases
Prion is a subclass of amyloid, which is constituted by protein aggregates that are self-perpetuating, self-propagating, and highly infectious, with the capability to cross-seed aggregation in other proteins [
93]. Prions and amyloids share a common biochemical basis. However, prions are infectious, whereas other amyloids are non-infectious in higher eukaryotes due to the ease of transmissibility of prion protein from one organism to another. Like amyloids, prions can be pathogenic or functional [
94]. Missense, insertion, and point mutations in the prion protein gene (
PRNP) cause abnormal production of the prion protein (PrP), which has a tendency to self-aggregate and accumulate as insoluble fibrils [
95].
Molecular mechanisms of PrP-seeded amyloid fibril formation have been deciphered with the help of CD, mass spectrometers, ultracentrifugation, and chemical cross-linking [
115]. The TEM study revealed that PrP rods are 10 to 20 nm in diameter and 100 to 200 nm in length in negative staining. By rotating with tungsten, the individual rods are 25 nm in diameter. Cryo-EM analysis evinces that each PrP fibril is composed of two protofibrils intertwined in a left-handed helix, and the fibril core diameter is ~14 nm with a width of ~25 nm [
116]. From a solid-state NMR study, it is evident that the PrP
Sc fibrils have parallel intermolecular β-sheet architectures [
117]. Like other amyloids, prion aggregates also reveal characteristic amyloid dye-binding properties since after binding with ThT, PrP amyloid fibrils exhibited an enhanced fluorescence at 485 nm and revealed apple-green birefringence under cross-polarized light after staining of fibrils with CR [
118]. A diagrammatic representation of amyloid formation in prion diseases is illustrated in
Figure 3 [
116,
119,
120,
121,
122].
Figure 3. A schematic representation of prion protein (PrP) fibril formation resulting in spongiform degeneration [
116,
119,
120,
121,
122].
PrPC: cellular form of PrP, and
PrPSC: infectious form of PrP.
6. IAPP Aggregation and Type 2 Diabetes
Islet amyloid polypeptide (IAPP) or amylin is a pancreatic hormone produced by pancreatic β-cells [
123]. Amylin is a 32-amino-acid-based peptide. Mutation in the islet amyloid polypeptide (
IAPP) gene results in the formation of amylin amyloid fibrils, which are primarily observed in late-onset type 2 diabetes (T2D) [
124]. In 1993, O’Brien et al. discussed the biological role of IAPP and its function in T2D [
125]. Lorenzo and coworkers investigated the toxicity of IAPP fibrils to the β-cells, and from their experiments, it was evident that human IAPP self-assemblies are toxic to islet β-cells [
126]. Howard described the deposition of toxic amylin aggregates in primates and cats and correlated those toxic assemblies with T2D in these mammals [
127]. Among these mammals, only rats could not produce amyloids, and as a result, they do not generate T2D [
128].
Westermark et al., for the first time, investigated the amyloid nature of IAPP aggregates using optical microscopy in T2D [
134]. EM, AFM, and X-ray fiber diffraction techniques showed that the structures of IAPP aggregates resemble twisted protofilaments with distinct cross-β sheets. EM of IAPP amyloid fibrils present that the fibrils are long, unbranched, and exhibit a diameter of about 100 Å. An X-ray diffraction pattern depicts that the peak positions of the IAPP fibril occur at 4.7 Å meridional and 10 Å equatorial reflections corresponding to a cross-β pattern. CD and FTIR studies further implicate that the IAPP aggregates predominantly contain β-sheets. The Cryo-EM analysis of full-length IAPP fibrils in ice revealed a strong reflection at 4.7 Å which is similar to a strong meridional signal at 4.7 Å observed for Aβ (11–25) fibrils, supporting this cryo-EM feature as a common characteristic present in amyloid fibrils [
135].
7. Amyloid-like Aggregation in the Patho-Physiology of Cancer
The p53 protein acts as a global transcription factor to maintain the integrity of the cells under different stresses [
144]. The p53 consists of 393 amino acid residues and abnormality in its biological function leads to cancer. The mutation in the tumor-suppressor gene (
TP53) causes the formation of abnormal p53 protein, which has a tendency to self-aggregate and form amyloidogenic structures [
145,
146]. Interestingly, the aggregation propensity of the p53 protein is remarkably like the PrP protein, and likewise, PrP and p53 aggregates are also infectious and cause cross-seed aggregation in other proteins [
146]. Maji and his team, through their studies, demonstrated that aggregation of p53 protein results in the impairment of its regular functions leading to the production of cells with leaky membranes, a characteristic feature of cancer pathogenesis [
147]. Several research findings around the globe suggest that almost half of human cancers are associated with p53 gene mutation [
145,
147]. The studies by De Oliveira and others suggest that the p53 DNA binding domain (DBD) is more prone to pressure-induced unfolding and aggregation due to poorer backbone hydrogen bond formation between p53 and DBD [
148]. Maji and coworkers examined the contribution of wild-type p53 aggregates in the formation of tumors and hypothesize its implications as potential therapeutic targets [
149].
8. Metabolites Assemblies as a Surprising Extension to Generic Amyloid Hypothesis
8.1. Amyloid-like Aggregation of Single Amino Acids and Its Implications in IEMs
Gazit and coworkers, for the very first time, delineated a common etiology between rare IEM phenylketonuria (PKU) and amyloid-associated diseases. In their pathbreaking research, the group reported amyloid-like fibrils formed by the self-assembly of phenylalanine (Phe). Mutation in the phenylalanine hydroxylase (
PAH) gene causes deficiency in the phenylalanine hydroxylase (PAH) enzyme. The blood concentration of Phe is elevated due to the deficient activity of the PAH enzyme. Elevated Phe concentration is responsible for autosomal recessive disorder PKU. The study revealed that Phe fibrils, just like conventional amyloid, could bind CR and ThT dye. The MTT assay suggests that Phe fibrils are cytotoxic to both hepatic as well as neural cell lines. The immune-histochemical analysis connotes the formation of antibodies against Phe fibrils. Hence, the results suggest a common etiology between PKU and amyloid-associated diseases and implicates that pathogenesis in PKU is associated with the formation of toxic fibrillar assemblies by phenylalanine [
17].
Subsequent to the Phe fibril discovery, Gazit and his team explained the abrupt formation of amyloid-like supramolecular nanostructures by the self-assembly of tyrosine (Tyr) [
20]. The rare IEM disorder tyrosinemia occurs due to the accumulation of Tyr in the body, which is also caused by a mutation in the gene-expressing enzyme tyrosine hydroxylase (TyH), which catabolizes Tyr [
156]. Ménard-Moyon et al. encountered that the Tyr amyloid nano-fibrillar structures accelerate the aggregation of globular proteins and aromatic metabolites, and this type of amyloid cross-seeding results in the creation of a lethal aggregation trap of proteins. For instance, tyrosine can self-assemble into a variety of structures, such as nanoribbons, dendritic structures, fiber-like structures, etc. [
157]. Tyr amyloid structures display an apple-green color after staining with CR and an enhanced fluorescence intensity after binding with ThT. The cytotoxicity analysis of Tyr assemblies by XTT assay suggests that upon increasing the concentrations of Tyr from 0.2 to 4 mg/mL, the cell viability was decreased from 80% to 40% [
20].
8.2. Non-Proteinaceous Metabolite Assemblies and Their Implications in IEMs
Gazit and coworkers have recently reported research on the association of homocysteine amyloid toxic fibrils with Alzheimer’s disease pathogenesis. They characterized the homocysteine crystals and their amyloid fibrils. Furthermore, they demonstrated the inhibition methodology of homocysteine fibrils by polyphenolic compounds [
165]. Zaguri et al. examined the self-assembly process of oxalate, which results in supramolecular nanofibrils without the accumulation of calcium. They provided mechanistic insight to explain the inconsistency between impaired retinal function and lack of crystal deposition by examining the self-assembly of oxalate into supramolecular nano architects. Using TEM analysis, they displayed the existence of elongated oxalate nano-fibrillar structures. They also obtained the crystals of oxalate by using a supersaturated solution containing ~70 mM of calcium oxalate in phosphate buffer saline (PBS) [
166]. Quinolinic acid (QA) is a neurometabolite in the kynurenine pathway, the biosynthetic pathway of tryptophan, which is associated with deadly neurodegenerative diseases.
Gazit and his team delineated the cytotoxicity of adenine, orotic acid, and uracil self-assemblies. Cytotoxicity of adenine was carried out using an XTT assay in SH-SY5Y cells, and it was found that as the concentration of adenine was increased to 2 mg/mL, cell viability decreased around 50%, which is similar to that of orotic acid. Uracil assemblies, on the other hand, displayed 90% cell viability at the same concentration. From the cytotoxicity analysis, it was surmised that adenine and orotic acid self-assemblies exhibit more cytotoxicity compared to uracil aggregates. On the other hand, the apoptotic behavior of adenine, orotic acid, and uracil assemblies was analyzed using annexin V and propidium iodide (PI) assays.
Gazit and coworkers recently reported robust twisted ribbon-like structures formed by the self-assembly of glucosylceramide (GlcCer). Notably, a deficiency of the lysosomal enzyme glucocerebrosidase results in the accumulation of GlcCer and causes Gaucher’s disease [
169]. The cytotoxicity analysis of GlcCer aggregates, along with other characteristic amyloidogenic traits, suggest that the etiology of Gaucher’s disease is also associated with amyloidosis [
169]. Kar and coworkers discussed amyloid-mimicking self-assemblies formed by artificial sweetener aspartame through conventional microscopy analysis, turbidimetry, ThT-binding assay cytotoxicity, and hemolysis analysis, along with MD-simulation experiments [
170].
9. Functional Amyloid
Amyloids are not always harmful, albeit they also play important functional roles in nature, like adherence to abiotic surfaces, resistance to antibiotics, detoxification of harmful compounds, assisting in electron transport, morphological differentiation of filamentous bacteria, and biofilm formation. Although the term amyloid is often associated with diseases, scientists have found various cases where amyloid fibrils play important functional roles in nature, from prokaryotic cells to mammals. Amyloids help to adhere to abiotic surfaces [
175], detox harmful compounds [
176], resist antibiotics [
177], direct morphological differentiation of filamentous bacteria [
178], and assist electron transport [
179].
Functional amyloids are also excellent candidates for the design of hydrogels. Natural amyloids formed by β-lactoglobulin and lysozyme proteins have been used for the fabrication of antibacterial hydrogels. The mechanical properties of amyloid fibrils are better than actin or tubulin, owing to their higher Young’s modulus of 3.3 ± 0.4 GPa and a peak tensile strength of 0.6 ± 0.4 GPa [
186,
187]. Hence, amyloid fibers are excellent scaffolds for the design of structural materials like free-standing films and fibers [
188,
189,
190]. Amyloid fibrils have also been used for the design of organic–inorganic hybrid nanomaterials owing to their high aspect ratios and the presence of multiple binding sites along their surface, which allows the fabrication of nanoscale amyloid-inorganic hybrid materials through post-assembly or co-assembly modification [
191].
10. Amyloid Cross-Seeding and Interaction
The formation of amyloid fibrils is regulated by the formation of “seeds”, which are stable nuclei that promote fibril formation by converting soluble proteins to fibrils. Amyloid fibrils formed by aggregation of specific proteins are often entangled and found in association with other proteins. For example, neurofibrillary tangles present in AD have both Ab and tau fibrils. Hence, cross-seeding or cross-talk between different amyloidogenic fibrils is a common phenomenon that may be homologous or heterologous [
192]. In homologous cross-seeding seeds of one type of protein interact and facilitate fibril formation, while in heterologous cross-seed, the seed of one type of protein facilitates the aggregation of another amyloid protein. Hence, more than one misfolded protein can cause pathogenesis in one disease, and more than one type of protein misfolding disease (PMD) can co-exist in an individual [
193,
194]. It has also been observed that individuals diagnosed with one PMD are more susceptible to developing another [
195,
196]. There is a plethora of literature reports wherein cross-seeding of known amyloidogenic proteins, namely Aβ42, Aβ40 PrP tau IAPP, and α-Syn, has been cross-seeded with each other, and the results illustrate that cross-seeding has a synergistic effect on the aggregation propensities and toxicities associated with individual amyloidogenic proteins [
197].
Cross-interactions might also have beneficial roles when the presence of other proteins inhibits the formation of toxic aggregates by major amyloid constituents. Some proteins, for example, TTR, CysC, and apoA-1, inhibit Aβ fibrillation, leading to delayed AD progression [
193]. Many small molecule inhibitors have also been efficiently identified by cross-seeding experiments. In this context, a small molecule, MG-2119, was screened as a potent inhibitor for both monomeric tau and α-Syn aggregation [
198]. The oligomerization and fibrillogenesis of both Aβ and α-Syn protein can be efficiently inhibited by anti-Parkinsonian drugs entacapone and tolcapone [
199]. Aggregation of Aβ and α-syn can also be inhibited by curcumin [
200]. Fibrillation of both Aβ and hIAPP can be inhibited by polyphenol pentagalloyl glucose (PGG) [
201,
202].
11. Critical Analysis and Future Outlook
The studies provide a comprehensive literature survey that implicates the validity of the generic amyloid hypothesis in the etiology of a plethora of diseases like AD, PD, prion diseases, T2D, cancer, and IEMs. Abundant evidence also suggests that common therapeutic remedies for these diseases can be envisaged by designing drugs that could act as generic amyloid inhibitors. However, it is apparent that the diseases like AD, which was first discovered to be caused by amyloid-like aggregation, still do not have a cure. This is surprising, and the reason for the inefficacy of these drugs is still unknown. Hence, if the generic amyloid hypothesis is considered a common cause for pathogenesis in other diseases, including the most recently discovered association with IEMs, its significance in unraveling the therapeutic remedy for these diseases is still questionable since a drug that inhibits such amyloid-like progression should have been clinically successful. However, the limited success of such drugs in AD implies that there might be another pathway through which the disease may progress. The clinical trials suggest that most drugs that could inhibit aggregation and hinder amyloid-like aggregation have not been found effective in the treatment of this disease. There should be a lot more clinical studies for assessing the effect of generic amyloid inhibitors like polyphenols, tannic acid, quercetin, and flavonoids as common therapeutic drugs for diseases to unravel the role of the amyloid hypothesis in their etiology. Considering the failures of previous drug trials, it is imperative to design a new drug or screen a potential FDA-approved drug that could arrest aggregation at the nucleation step of amyloid growth. One possible reason for drug failures might be a lack of understanding of therapeutic targets or identification of wrong pathological substrates. Hence, it is imperative to pursue more research on the ultrastructure of different amyloids and understand the mode of their propagation and transmission.
This entry is adapted from the peer-reviewed paper 10.3390/life13071523