Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Siddhant Bhoir | -- | 1911 | 2023-07-10 16:04:14 | | | |
| 2 | Fanny Huang | Meta information modification | 1911 | 2023-07-11 08:17:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bhoir, S.; De Benedetti, A. Clinical Signs of Androgen Receptor-Negative Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/46607 (accessed on 28 July 2026).
Bhoir S, De Benedetti A. Clinical Signs of Androgen Receptor-Negative Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/46607. Accessed July 28, 2026.
Bhoir, Siddhant, Arrigo De Benedetti. "Clinical Signs of Androgen Receptor-Negative Disease" Encyclopedia, https://encyclopedia.pub/entry/46607 (accessed July 28, 2026).
Bhoir, S., & De Benedetti, A. (2023, July 10). Clinical Signs of Androgen Receptor-Negative Disease. In Encyclopedia. https://encyclopedia.pub/entry/46607
Bhoir, Siddhant and Arrigo De Benedetti. "Clinical Signs of Androgen Receptor-Negative Disease." Encyclopedia. Web. 10 July, 2023.
Copy Citation
Androgen deprivation therapy (ADT) has been the mainstay of prostate cancer (PCa) treatment, with success in developing more effective inhibitors of androgen synthesis and antiandrogens in clinical practice. However, hormone deprivation and AR ablation have caused an increase in ADT-insensitive PCas associated with a poor prognosis. Resistance to ADT arises through various mechanisms, and most castration-resistant PCas still rely on the androgen axis, while others become truly androgen receptor (AR)-independent.
PCa
AR
ADT
TLK1 Signaling
1. Introduction
Androgen steroid hormones play a crucial role in PCa by binding to the AR and triggering a specific oncogenic transcriptional program [1]. This understanding has been exploited for years to manage the disease’s initial or recurrent metastatic spread after surgery or radiotherapy. Despite the temporary effectiveness of hormone-deprivation therapies and anti-androgens in halting tumor growth, most patients eventually develop resistance and progress to the incurable phase of mCRPC [2][3][4][5] (Figure 1). However, targeting PCa cells early before progressing to mCRPC can significantly improve outcomes [6] (Figure 1). This can be achieved by the selective intervention of the AR-dependent and independent compensatory pathways that drive mCRPC development [7][8][9].
Recent more potent inhibitors of AR signaling (abiraterone, enzalutamide, apalutamide, and darolutamide) demonstrate some benefits for castration-resistant PCa (CRPC) patients, highlighting the critical role of AR signaling [10]. CRPC cells adapt to low androgen levels through AR gene mutations and amplifications [11], resulting in constitutively active splice variants that directly regulate the expression of DNA repair genes [12][13][14][15][16]. In addition, the upregulation of coactivators [17][18] and androgen-producing enzymes within the tumor contributes to AR signaling persistence in CRPC, making the disease drug refractory [16][19]. The complex and multifaceted nature of the mechanisms driving CRPC development and progression makes it challenging to identify effective therapies. Consequently, searching for successful CRPC treatments continues to pose a significant obstacle in PCa research.
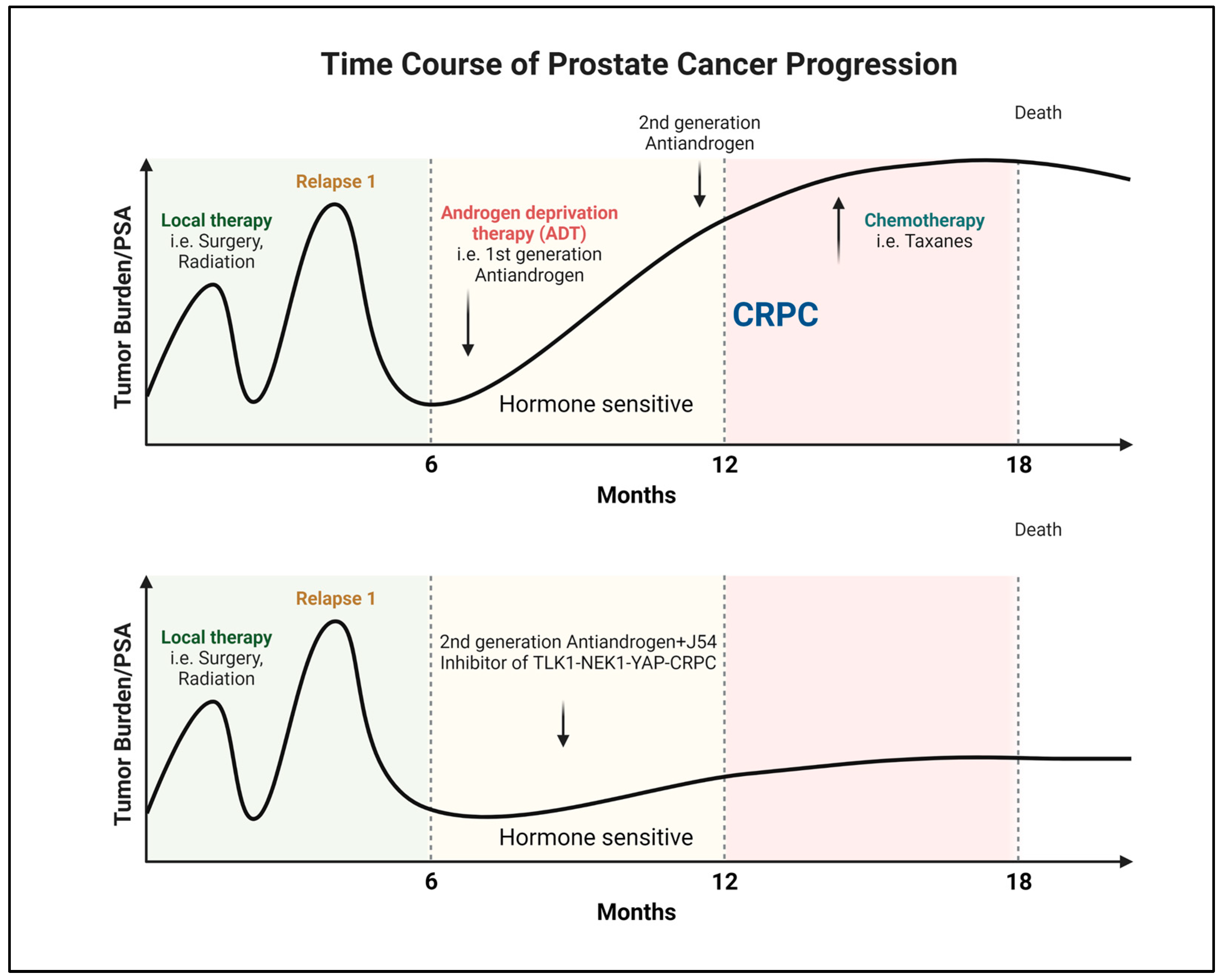
Figure 1. Progression and recurrence of prostate cancer measured biochemically (PSA). Created with BioRender.com.
In recent years, researchers' laboratory has discovered that the DNA damage and response (DDR) kinase TLK1 plays a vital role in facilitating the adaptation of PCa cells to ADT (Figure 2). Initially, TLK1 promotes a cell cycle arrest by activating the TLK1-NEK1-ATR-Chk1 kinase cascade, preventing PCa cells from entering the cell cycle when faced with unfavorable growth conditions during androgen deprivation [20][21][22][23]. This arrest is a protective measure to halt replication due to the lack of androgen. Furthermore, TLK1 helps reprogram PCa cells to adapt to androgen-independent growth, which is crucial for developing CRPC. This reprogramming occurs through the NEK1-YAP/AR-CRPC conversion pathway, allowing the cells to adapt and survive with limited androgen stimulation [24][25]. TLK1 also exhibits a significant anti-apoptotic role by regulating the NEK1-VDAC1 pathway, which modulates intrinsic mitochondrial apoptotic signaling when the DDR is activated [26]. This regulation helps prevent cell death and enhances the survival of PCa cells. Additionally, researchers' recent findings have revealed the essential role of TLK1 in promoting motility and metastasis. TLK1 indirectly regulates the kinases MK5/PRAK and AKT’s activity through AKTIP [27]. These regulatory mechanisms contribute to the increased motility and metastatic potential of PCa cells and therapy resistance.
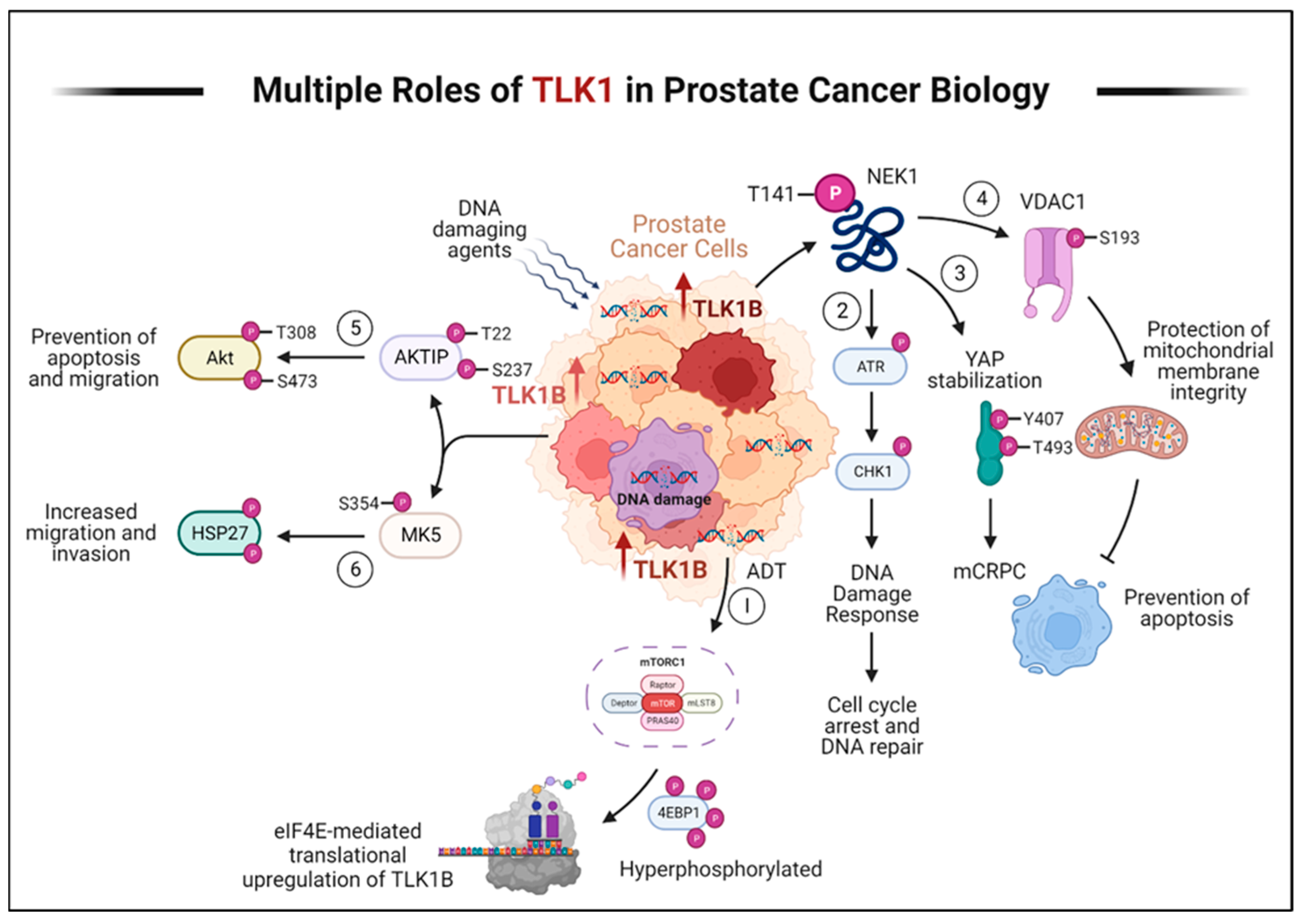
Figure 2. TLK1 plays multiple crucial roles in prostate cancer progression and therapy resistance. Here, the various mechanisms by which TLK1 contributes to these processes are described. (1) ADT triggers the activation of mTORC1, which activates 4EBP1, causing the release of eIF4E. Excess eIF4E initiates the translation of TLK1B. (2) TLK1/1B, once produced, activates NEK1 by phosphorylating it at T141. Activated NEK1 activates the ATR-Chk1 DDR signaling cascade. The activation of DDR promotes DNA repair, which aids in the resistance to DNA-damaging therapeutic agents. (3) Through TLK1-NEK1 signaling, YAP is phosphorylated on Y407 and T493 residues, stabilizing it. This phosphorylated YAP binds to TEAD or other transcription factors (TF) and relocates to the nucleus, evading proteasomal degradation. The accumulation and stabilization of YAP contribute to the progression of CRPC and resistance to drugs. (4) The TLK1-NEK1 axis also plays a role in phosphorylating VDAC1 at S193, which helps maintain the integrity of the mitochondrial membrane and inhibits intrinsic apoptotic signaling. (5) TLK1 directly phosphorylates AKTIP on T22 and S237 residues, activating AKT. This activation of AKT promotes pro-survival and pro-migratory signaling. (6) TLK1 also interacts with and phosphorylates MK5, enhancing its catalytic activity towards HSP27, a substrate of MK5. This increased activity of MK5 leads to enhanced prostate cancer cell migration, invasion, and metastasis. Created with BioRender.com.
Recognizing TLK1 as a pivotal contributor to PCa’s ability to adapt to ADT, promote growth with minimal androgen stimulation, evade apoptosis, and facilitate cell motility and metastasis offers a hopeful foundation for the ongoing quest for potential treatments. By focusing on TLK1 and its related pathways, researchers can develop innovative strategies to delay or halt the progression of PCa to the incurable mCRPC stage. Therefore, researchers' endeavor to comprehend the biology of TLK1 represents a particular initiative to comprehensively understand PCa and identify appropriate therapeutic options for clinical trials.
2. Progression of PCa to AR-Negative Lethal Disease: Understanding the Implications
Recent advancements in inhibiting AR signaling, mainly through potent AR-targeted therapies, have resulted in a concerning trend of metastatic PCas transforming into AR-negative diseases. This change renders these tumors unresponsive to inhibition of the AR signaling pathway. While AR-negative disease is uncommon in untreated patients, its incidence has been rising in individuals receiving AR-targeted therapies [28], a trend expected to continue as these treatments become more widespread across various stages of the disease. A study conducted by Bluemn et al. revealed that the proportion of AR-negative tumors in patients with mCRPC significantly increased from 11% (1998–2011) to 36% (2012–2016) following the introduction of potent AR-targeted therapies like enzalutamide and abiraterone [28]. Although there is currently no available data, it is plausible that even the utilization of highly effective small-molecule AR degraders entering clinical trials could further elevate the percentage of AR-negative mCRPC [29][30][31].
Treatment options for AR-negative PCa, specifically the subtype with neuroendocrine differentiation, are still uncertain due to a lack of consensus. Diagnosing therapy-induced AR-negative disease is challenging as it requires assessing current tumor tissue. The recommended approach for confirmed AR-negative disease with neuroendocrine differentiation is a platinum-based regimen, similar to treatments for other neuroendocrine small cell carcinomas. However, the response rates to combinations of cisplatin and carboplatin with docetaxel or etoposide are relatively high but not long-lasting [32][33][34]. As a result, the prognosis for patients is poor, with average survival ranging from 12 to 36 months [33][35][36]. Therefore, it has become crucial to comprehend the occurrence of AR-negative PCa, which presents an urgent clinical need in the field.
3. Clinical Signs of AR-Negative Disease
During the progression of PCa, the loss of AR expression is accompanied by significant alterations in cellular differentiation, forming part of a more extensive cellular rewiring process [37]. This shift leads to the emergence of diverse subtypes of AR-negative PCas, each displaying distinct cellular characteristics. Identifying and distinguishing these subtypes based on morphological and molecular traits can be challenging, as some tumors may exhibit different features and gradual changes within a single nodule [38]. Consequently, the classification of AR-negative PCas remains a significant hurdle in the field.
Neuroendocrine PCa (NEPC), also known as small cell PCa, is a significant subtype among AR-negative tumors [38][39]. Neuroendocrine and basal-like proteins characterize it, while the expression of luminal and epithelial markers regulated by the AR is diminished [38]. NEPC exhibits a spectrum of histological features, ranging from well-differentiated neuroendocrine tumors to highly aggressive cancers with small cell morphology [40]. Interestingly, there are also rare histological subtypes that show squamous differentiation [41].
Newer functional analyses have demonstrated that NEPC typically originates from an AR-positive adenocarcinoma through transdifferentiation. Zou et al., in lineage-tracing mouse model studies, showed that the neuroendocrine features arise from the transdifferentiation of luminal cells [42]. Inactivating the p53 gene increased the expression of neuroendocrine markers and reduced the response to abiraterone. Using a YFP tracer under the control of the Nkx3.1 luminal-specific promoter, the study revealed that almost all tumors with neuroendocrine markers also expressed YFP, providing evidence of their luminal-epithelial origin. Additionally, androgen-sensitive prostate adenocarcinoma cells have been observed to undergo neuroendocrine differentiation in an androgen-depleted cell medium, suggesting that castration actively promotes the development of NEPC [43]. The emergence of the neuroendocrine phenotype is partly due to the suppression of AR expression, as AR increases the expression of the neuronal transcription factor BRN2 [44]. AR can directly suppress BRN2 expression, and BRN2 can modulate the activity of SOX2, a key driver of cellular plasticity [44]. Recent cancer genomics studies have also supported transdifferentiation, as NEPC often exhibits genetic alterations reminiscent of AR-dependent CRPC. These include highly recurrent AR mutations and TMPRSS2-ERG gene fusions [36][45][46][47]. It is worth noting that these TMPRSS2-ERG gene rearrangements activate the transcriptional program regulated by YAP1 and that prostate-specific activation of either ERG or YAP1 in mice induces similar transcriptional changes and results in age-related prostate tumors [24][48][49]. These findings further emphasize the significance of targeting the TLK1-NEK1-YAP pathway during early adaptation to ADT.
Recent findings have revealed a connection between the activation of Wnt/β-Catenin signaling and the presence of AR-negative disease. This connection is established through the involvement of specific genes regulated by Wnt/β-Catenin, namely FOXA2, and MYCN, which promote the process of neuroendocrine transdifferentiation [50][51][52][53][54]. Laboratory experiments conducted on PCa cells have indicated that the presence of active β-Catenin leads to an increase in the production of neuroendocrine-specific proteins like NSE and chromogranin A [55]. Interestingly, inhibiting Wnt/β-Catenin signaling has decreased neuroendocrine transdifferentiation in laboratory settings [55]. Additionally, the upregulation of Wnt-11 has been observed in NEPC, and it has been functionally linked to the transdifferentiation process in vitro [56].
A recently identified subtype of AR-negative PCa, the double-negative subtype, has become more prevalent in recent years. This subtype, which lacks AR and neuroendocrine markers, has shown a strong association with the use of newer AR pathway inhibitors [28]. Its occurrence has increased from 5% to over 20% in the past decade, making it one of the most common types of AR-negative PCa [28]. The growth of these double-negative tumors is supported by heightened autocrine FGF signaling, which activates the MAPK pathway and contributes to their proliferation [28]. Consequently, these cancer cells exhibit sensitivity to pharmacological inhibition of FGF and MAPK signaling.
A newly discovered subtype of mCRPC has been linked to the loss of chromodomain helicase DNA-binding protein 1 (CHD1). When CHD1 is deficient, cancer cells undergo significant changes in their chromatin structure, making them resistant to enzalutamide [57]. These resistant tumors often exhibit higher glucocorticoid receptor (GR) expression, leading to sustained signaling that promotes drug resistance. However, inhibiting GR in the presence of CHD1 deficiency can restore sensitivity to enzalutamide [57], indicating that increased GR activity is crucial in developing resistance to this drug.
Labrecque et al. recently identified five mCRPC subtypes based on AR and neuroendocrine marker RNA expression patterns. These subtypes include tumors with high AR expression (ARPC), low AR expression (ARLPC), tumors expressing both AR and neuroendocrine markers (AMPC), tumors lacking both AR and neuroendocrine markers (DNPC), and tumors exhibiting small cell and neuroendocrine features without AR expression (SCNPC) [58]. Although progress has been made, further research is needed to understand the similarities and differences among these AR-negative subtypes regarding clinical characteristics and potential treatment options. This will require collaborative efforts from multidisciplinary teams, the integration of molecular biomarkers, and precise patient selection, as highlighted in a recent workshop sponsored by the National Cancer Institute (NCI) [59].
The subtypes mentioned earlier are only sometimes distinct from each other and can change over time [58]. They likely represent stages of a continuous process where cancer cells lose their specialized characteristics, acquire stem cell-like properties, and eventually become independent of and resistant to AR signaling [58][60][61].
References
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308.
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85.
- Merseburger, A.S.; Haas, G.P.; von Klot, C.A. An update on enzalutamide in the treatment of prostate cancer. Ther. Adv. Urol. 2015, 7, 9–21.
- Kumar, J.; Jazayeri, S.B.; Gautam, S.; Norez, D.; Alam, M.U.; Tanneru, K.; Bazargani, S.; Costa, J.; Bandyk, M.; Ganapathi, H.P.; et al. Comparative efficacy of apalutamide darolutamide and enzalutamide for treatment of non-metastatic castrate-resistant prostate cancer: A systematic review and network meta-analysis. Urol. Oncol. 2020, 38, 826–834.
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511.
- Imamura, Y.; Sadar, M.D. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int. J. Urol. 2016, 23, 654–665.
- Zhu, M.L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849.
- Craft, N.; Shostak, Y.; Carey, M.; Sawyers, C.L. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat. Med. 1999, 5, 280–285.
- Seruga, B.; Ocana, A.; Tannock, I.F. Drug resistance in metastatic castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 12–23.
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474.
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711.
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37.
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271.
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479.
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253.
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380.
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22.
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858.
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454.
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926.
- Polci, R.; Peng, A.; Chen, P.L.; Riley, D.J.; Chen, Y. NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res. 2004, 64, 8800–8803.
- Chen, Y.; Chen, P.L.; Chen, C.F.; Jiang, X.; Riley, D.J. Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201.
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180.
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666.
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126.
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375.
- Khalil, M.I.; Madere, C.; Ghosh, I.; Adam, R.M.; De Benedetti, A. Interaction of TLK1 and AKTIP as a Potential Regulator of AKT Activation in Castration-Resistant Prostate Cancer Progression. Pathophysiology 2021, 28, 339–354.
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e476.
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119.
- Flanagan, J.J.; Neklesa, T.K. Targeting Nuclear Receptors with PROTAC degraders. Mol. Cell Endocrinol. 2019, 493, 110452.
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; A Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110, An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259.
- Fléchon, A.; Pouessel, D.; Ferlay, C.; Perol, D.; Beuzeboc, P.; Gravis, G.; Joly, F.; Oudard, S.; Deplanque, G.; Zanetta, S.; et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann. Oncol. 2011, 22, 2476–2481.
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.-M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630.
- Loriot, Y.; Massard, C.; Gross-Goupil, M.; Di Palma, M.; Escudier, B.; Bossi, A.; Fizazi, K. Combining carboplatin and etoposide in docetaxel-pretreated patients with castration-resistant prostate cancer: A prospective study evaluating also neuroendocrine features. Ann. Oncol. 2009, 20, 703–708.
- Metzger, A.L.; Abel, S.; Wegner, R.E.; Fuhrer, R.; Mao, S.; Miller, R.; Beriwal, S.; Horne, Z.D. Patterns of care and outcomes in small cell carcinoma of the prostate: A national cancer database analysis. Prostate 2019, 79, 1457–1461.
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18.
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; MacVicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216.
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850.
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436.
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.-M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767.
- Randolph, T.L.; Amin, M.B.; Ro, J.Y.; Ayala, A.G. Histologic variants of adenocarcinoma and other carcinomas of prostate: Pathologic criteria and clinical significance. Mod. Pathol. 1997, 10, 612–629.
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749.
- Zhang, X.Q.; Kondrikov, D.; Yuan, T.C.; Lin, F.F.; Hansen, J.; Lin, M.F. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene 2003, 22, 6704–6716.
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71.
- Lotan, T.L.; Gupta, N.S.; Wang, W.; Toubaji, A.; Haffner, M.C.; Chaux, A.; Hicks, J.L.; Meeker, A.K.; Bieberich, C.J.; De Marzo, A.M.; et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod. Pathol. 2011, 24, 820–828.
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495.
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305.
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell 2015, 27, 797–808.
- Ghosh, I.; Khalil, M.I.; Mirza, R.; King, J.; Olatunde, D.; De Benedetti, A. NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression. Biomedicines 2023, 11, 743.
- Yu, X.; Wang, Y.; Jiang, M.; Bierie, B.; Roy-Burman, P.; Shen, M.M.; Taketo, M.M.; Wills, M.; Matusik, R.J. Activation of beta-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate 2009, 69, 249–262.
- Kuwahara, A.; Hirabayashi, Y.; Knoepfler, P.S.; Taketo, M.M.; Sakai, J.; Kodama, T.; Gotoh, Y. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development 2010, 137, 1035–1044.
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 129, 3924–3940.
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577.
- Park, J.W.; Lee, J.K.; Witte, O.N.; Huang, J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol. 2017, 30, 1262–1272.
- Yang, X.; Chen, M.-W.; Terry, S.; Vacherot, F.; Chopin, D.K.; Bemis, D.L.; Kitajewski, J.; Benson, M.C.; Guo, Y.; Buttyan, R. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res. 2005, 65, 5263–5271.
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010, 9, 55.
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e511.
- Labrecque, M.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; Gil Da Costa, R.M.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505.
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924.
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503.
- Alumkal, J.J.; Sun, D.; Lu, E.; Beer, T.M.; Thomas, G.V.; Latour, E.; Aggarwal, R.; Cetnar, J.; Ryan, C.J.; Tabatabaei, S.; et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 12315–12323.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
709
Revisions:
2 times
(View History)
Update Date:
11 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No