Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Po-Wah So | -- | 1666 | 2023-06-30 14:19:16 | | | |
| 2 | Wendy Huang | Meta information modification | 1666 | 2023-07-03 05:09:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Inyang, D.; Saumtally, T.; Nnadi, C.N.; Devi, S.; So, P. Effects of Capsaicin on Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/46276 (accessed on 23 July 2026).
Inyang D, Saumtally T, Nnadi CN, Devi S, So P. Effects of Capsaicin on Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/46276. Accessed July 23, 2026.
Inyang, Deborah, Tasneem Saumtally, Chinelo Nonyerem Nnadi, Sharmila Devi, Po-Wah So. "Effects of Capsaicin on Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/46276 (accessed July 23, 2026).
Inyang, D., Saumtally, T., Nnadi, C.N., Devi, S., & So, P. (2023, June 30). Effects of Capsaicin on Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/46276
Inyang, Deborah, et al. "Effects of Capsaicin on Alzheimer’s Disease." Encyclopedia. Web. 30 June, 2023.
Copy Citation
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder characterised by cognitive impairment, and amyloid-β plaques and neurofibrillary tau tangles at neuropathology. Capsaicin is a spicy-tasting compound found in chili peppers, with anti-inflammatory, antioxidant, and possible neuroprotective properties. Capsaicin intake has been associated with greater cognitive function in humans, and attenuating aberrant tau hyperphosphorylation in a rat model of AD.
capsaicin
Alzheimer’s Disease
cognition
amyloid
tau
1. Introduction
Dementia is estimated to affect 55 million people globally and this number is expected to rise to 139 million by 2050, due to the ageing global population [1]. Alzheimer’s Disease (AD), the most common cause of dementia, accounts for 60–70% cases [1][2]. AD is a progressive neurodegenerative disorder, characterised by cognitive impairment, neuronal loss (brain atrophy), and the accumulation of amyloid-β (Aβ) plaques and neurofibrillary tau tangles (NFTs)—the latter abnormal protein aggregates being neuropathological hallmarks of AD. Memory impairments are typically the first sign of AD, specifically episodic memory, followed by impairments in declarative memory and non-declarative memory [3][4]. In addition to memory impairments, other cognitive domains such as attention, executive functioning, language, perceptual-motor function, social cognition, and the inability to perform the activities of daily living, form the basis of diagnosis of dementia [4].
AD can be broadly categorised into two types [5], although rarer forms of atypical AD exist. Sporadic, or late-onset AD, occurs mostly in patients > 65 years of age, and accounts for nearly 95% of AD cases [5]. Early-onset AD affects those aged <60 years of age, with 60% having familial AD (FAD), and accounts for 1–5% of AD cases [6]. FAD arises from a mutation of the genes that encode the Amyloid Precursor Protein (APP) and presenilin (PSEN1 and PSEN2), which are involved in APP breakdown and Aβ generation. Late- and early-onset AD are clinically indistinguishable, aside from differences in disease onset, with early-onset AD generally associated with more rapid progression and a Mendelian pattern of inheritance [6]. Atypical AD accounts for 5% of late-onset AD cases, and up to one-third of early-onset AD cases [7]. There are four types of atypical AD, including logopenic aphasia, which primarily affects language; posterior cortical atrophy, characterised by visuospatial/visuoperceptual disturbances; frontal variant AD, characterised by executive or behavioural-predominant dysfunction, similar to behavioural variant frontotemporal dementia; and corticobasal syndrome, characterised by motor disturbances [7]. Approximately 8–13% of AD patients present with motor or visual problems, 7–9% with language problems, and 2% with executive dysfunction [8][9]. Atypical forms of AD were previously considered separate conditions to AD; however, cerebrospinal fluid (CSF) and PET biomarkers of AD pathologies, such as CSF Aβ1-42 and PET tau, have shown a significant pathology overlap between AD and atypical forms [7][8], with 67–100% estimated to be logopenic aphasia cases, 76–100% primary cortical atrophy, 7–20% behavioural variant FTD, and 15–50% corticobasal syndrome. The APOE gene encodes apolipoprotein E that transports cholesterol [10]. APOE(ε4), the strongest genetic risk factor for AD is also a risk factor for developing posterior cortical atrophy and frontal variant AD [7]. Case studies suggest that PSEN1 mutations are a risk factor for developing corticobasal syndrome [11], whilst mutations in PSEN2 have been seen in posterior cortical atrophy [12].
AD progresses through a continuum comprising preclinical mild cognitive impairment (MCI) and AD [13][14]. The preclinical stage can be decades before symptoms are evident, including brain accumulation of extracellular Aβ-containing plaques and intracellular NFTs. The lack of symptoms means that biomarkers are crucial to identifying this stage. Biomarkers include CSF Aβ1-42 [15], Aβ1-42 being one of the two major isoforms of Aβ, and the major component of Aβ plaques [16]. MCI is a prodromal AD phase, characterised by cognitive deficits, particularly in memory, greater than that expected for normal ageing but without sufficient impairment to be diagnosed as AD [17]. MCI is associated with a higher risk of progression to clinically probable AD [13][14][18], with an annual conversion rate from MCI to AD reported to be 10–15% [19]. Thus, MCI is suggested to be a crucial stage at which the AD pathological processes are reversible, and provides a window of opportunity for greater therapeutic success.
Both Aβ and hyperphosphorylated tau protein are involved in AD pathogenesis, although their respective impacts are debated [20][21]. The amyloid hypothesis is currently the most widely accepted hypothesis for the pathogenesis of AD. It proposes that brain accumulation and aggregation of Aβ peptides resulting from abnormal APP processing is the main cause of AD [22]. APP can be processed via the non-amyloidogenic pathway (cleaved by α-secretase) or the amyloidogenic pathway (cleaved by β-secretase) (Figure 1). The amyloidogenic pathway leads to a build-up of Aβ plaques, contributing to neuroinflammation and subsequent cell death. Aβ peptides can also deposit around the cortical and leptomeningeal vasculature, which is termed cerebral amyloid angiopathy, contributing to microvascular dysfunction, neurovascular unit disintegration, and dysfunction of the blood–brain barrier [23][24]. According to the AD ‘vascular hypothesis’, Aβ peptide-associated vascular disruption is thought to contribute to neuroinflammation and chronic hypoperfusion, leading to impaired Aβ clearance and possibly triggering increased cerebrovascular Aβ production and aggregation [23]. Although the extent of vascular disruption in AD pathogenesis is unclear, hypertension and diabetes have been shown to significantly increase the risk of developing AD [25].
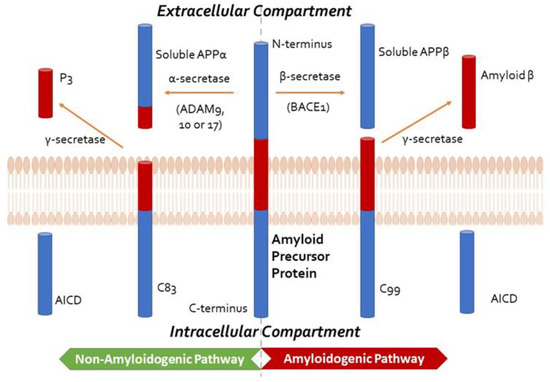
Figure 1. Summary of Amyloid Precursor Protein (APP) processing, depicting the non-amyloidogenic and the amyloidogenic pathways, the former precluding the formation of amyloid-β. In the non-amyloidogenic pathway, APP is cleaved by an α-secretase from the A disintegrin and metalloprotease (ADAM) family, such as ADAM9, ADAM10, and ADAM17, to form soluble APPα and C83. The smaller carboxy-terminal fragment, C83, can be cleaved by γ-secretase to generate P3 and the APP intracellular domain (AICD, not shown). In the amyloidogenic pathway, APP is cleaved by β-secretase (β-site APP-cleaving enzyme 1, BACE1) to produce soluble APPβ, retaining the last 99 amino acids of APP (known as C99) within the membrane. The peptide C99 is then cleaved by the γ-secretase complex, comprising presenilin 1 or 2, nicastrin, anterior pharynx defective-1 (APH-1), and presenilin enhancer 2 (PEN2) at the amino terminus to form amyloid-β and AICD. This cleavage predominantly produces Aβ1-40, and the more amyloidogenic Aβ1-42 at a ratio of 10:1.
Tau, a microtubule associated protein, maintains the structural stability of microtubules for cytoskeletal trafficking and organisation [26]. In AD, tau becomes abnormally hyper-phosphorylated, leading to microtubule disassembly. Free phosphorylated tau molecules are thought to aggregate into paired insoluble helical filaments, which accumulate and form NFTs [27]. Abnormal tau hyperphosphorylation is thought to be initiated by Aβ accumulation in AD [27]. In AD, NFT development appears to evolve with a predictable and hierarchical distribution pattern that starts from the entorhinal cortex, via the limbic system to the hippocampus and neocortex [28], although recent tau-PET data suggests tau pathology development is more heterogenous than originally thought [29]. Other AD pathological processes include synaptic dysfunction and loss [30][31], increased oxidative stress [32][33][34], neuroinflammation [35][36], electrophysiological abnormalities [37], and ultimately neuronal death, including via apoptosis [38]. Oxidative stress occurs early in AD, attributed to enhanced free radical overproduction, combined with insufficient antioxidant defence [5][33][39], such that reduced glutathione levels are observed in AD [33]. At the macroscopic level, hippocampal and neocortical atrophy with ventricular enlargement is observed [17].
AD currently has no cure and existing treatments have limited effects on attenuating disease progression [40]. The recent FDA approval of aducanumab is controversial, as, while aducanumab removes brain Aβ, its ability to improve cognition is debatable [41]. However, a recent report on another Aβ antibody, lecanemab, showed it was able to moderately lessen cognitive decline in early AD [42]. Thus, there is still an impetus to discover AD-modifying therapies. Notably, dementia is associated with increased polypharmacy, likely due to the management of associated comorbidities [43] that are, in turn, associated with an increased risk of negative clinical consequences [44]. Thus, there is much interest in exploring bioactives to treat AD.
2. Potential of Capsaicin in the Treatment of Alzheimer’s Disease
Capsaicin (8-methyl-N-vanillyl-6-nonenamide), a transient receptor potential vanilloid 1 (TRPV1)-receptor agonist, is a spicy-tasting, hydrophobic chemical found in most plants from the Capsicum genus [45][46]. There are over 20 different species of Capsicum (C), five of which have been domesticated: C. annuum, C. baccatum, C. frutescens, C. chinense, and C. pubescens [47]. Capsaicin concentrations vary depending on the plant species, ranging from 0.1 mg/g (C. annuum) to 60 mg/g (C. chinense), and accounts for ~71% of total capsaicinoids in most capsicum varieties [48]. Capsicum is often in human diets, for its flavour and spiciness. Estimates of global daily capsaicin consumption vary from 1.5 mg/person/day in the United States and Europe [49], to 25 mg/person/day in India, and 200 mg/person/day in Mexico [48]. The oral availability of capsaicin is 50–90% in animal studies [47]. Animal studies have shown capsaicin crosses the blood–brain-barrier [47], which is essential if it is to be considered as an AD therapy.
Physiologically, capsaicin is known for its ability to cause pain and the sensitisation of both peripheral and central nerves (leading to symptoms mimicking neuropathic pain, such as allodynia, secondary hyperalgesia, referred pain area, and visceral hyperalgesia). It is less well-known that capsaicin can induce desensitisation and the withdrawal of epidermal nerve fibres. The effects of capsaicin are dependent on the dose and route of administration [50].
Chilli peppers have been used for a broad range of therapeutic applications in Indian, Native American, African, and Chinese medicinal traditions for the treatment of rheumatism, arthritis, stomach ache, dog/snake bites, skin rashes, and wounds [51]. Commonly used as a topical analgesic [50], capsaicin has anti-inflammatory [52][53] and antioxidant effects [54][55], and it has also been shown to be neuroprotective against oxidative stress and apoptosis in epilepsy and ischaemic injury [56][57]. Recently, capsaicin was shown to attenuate hippocampal tau hyperphosphorylation in Type 2 diabetes rats injected with a streptozocin AD model [58]. Improved cognitive function was associated with spicy food consumption in humans [59][60], and lower CSF phospho-tau/Aβ1-42 and total tau/Aβ1-42 ratios in non-AD participants [60]. However, reports on its effects on amyloid levels are conflicting, as studies have shown decreased serum Aβ1-40 levels, total serum Aβ levels [59], and increased CSF Aβ1-42 levels [60]. There is currently no consensus on the recommended consumption to confer neuroprotective effects in humans. In a 15 year-long open cohort study, Shi et al., 2019, found adults with an average chilli consumption of 1–20 g/day had better cognitive scores than non-consumers [61]. However, those who consumed >50 g/day were more likely to have worse cognition, suggesting effects are dose-dependent [61].
References
- WHO. Fact Sheets of Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 25 May 2023).
- Gauthier, S.; Webster, C.; Servaes, S.; Morais, J.A.; Rosa-Neto, P. World Alzheimer Report 2022: Life after Diagnosis: Navigating Treatment, Care and Support; Alzheimer’s Disease International: London, UK, 2022.
- Gotz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598.
- Commins, S.; Kirby, B.P. The complexities of behavioural assessment in neurodegenerative disorders: A focus on Alzheimer’s disease. Pharmacol. Res. 2019, 147, 104363.
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of beta-amyloid peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311.
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512.
- Shea, Y.-F.; Pan, Y.; Mak, H.K.-F.; Bao, Y.; Lee, S.-C.; Chiu, P.K.-C.; Chan, H.-W.F. A systematic review of atypical Alzheimer’s disease including behavioural and psychological symptoms. Psychogeriatrics 2021, 21, 396–406.
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234.
- Koedam, E.L.; Lauffer, V.; van der Vlies, A.E.; van der Flier, W.M.; Scheltens, P.; Pijnenburg, Y.A. Early-versus late-onset Alzheimer’s disease: More than age alone. J. Alzheimers Dis. 2010, 19, 1401–1408.
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118.
- Lam, B.; Khan, A.; Keith, J.; Rogaeva, E.; Bilbao, J.; St George-Hyslop, P.; Ghani, M.; Freedman, M.; Stuss, D.T.; Chow, T.; et al. Characterizing familial corticobasal syndrome due to Alzheimer’s disease pathology and PSEN1 mutations. Alzheimers Dement. 2017, 13, 520–530.
- Carrasquillo, M.M.; Barber, I.; Lincoln, S.J.; Murray, M.E.; Camsari, G.B.; Khan, Q.U.A.; Nguyen, T.; Ma, L.; Bisceglio, G.D.; Crook, J.E.; et al. Evaluating pathogenic dementia variants in posterior cortical atrophy. Neurobiol. Aging 2016, 37, 38–44.
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279.
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562.
- Miller, D.L.; Papayannopoulos, I.A.; Styles, J.; Bobin, S.A.; Lin, Y.Y.; Biemann, K.; Iqbal, K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch. Biochem. Biophys. 1993, 301, 41–52.
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269.
- Tabuas-Pereira, M.; Baldeiras, I.; Duro, D.; Santiago, B.; Ribeiro, M.H.; Leitao, M.J.; Oliveira, C.; Santana, I. Prognosis of Early-Onset vs. Late-Onset Mild Cognitive Impairment: Comparison of Conversion Rates and Its Predictors. Geriatrics 2016, 1, 11.
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25.
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508.
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356.
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022.
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular Hypothesis of Alzheimer Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283.
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204.
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524.
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881.
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016, 12, 633–644.
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464.
- Di Domenico, F.; Pupo, G.; Giraldo, E.; Badìa, M.C.; Monllor, P.; Lloret, A.; Schininà, M.E.; Giorgi, A.; Cini, C.; Tramutola, A.; et al. Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic. Biol. Med. 2016, 91, 1–9.
- Chen, J.J.; Thiyagarajah, M.; Song, J.; Chen, C.; Herrmann, N.; Gallagher, D.; Rapoport, M.J.; Black, S.E.; Ramirez, J.; Andreazza, A.C.; et al. Altered central and blood glutathione in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 23.
- Peña-Bautista, C.; Baquero, M.; Vento, M.; Cháfer-Pericás, C. Free radicals in Alzheimer’s disease: Lipid peroxidation biomarkers. Clin. Chim. Acta 2019, 491, 85–90.
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. 2018, 4, 575–590.
- Walters, A.; Phillips, E.; Zheng, R.; Biju, M.; Kuruvilla, T. Evidence for neuroinflammation in Alzheimer’s disease. Prog. Neurol. Psychiatry 2016, 20, 25–31.
- López-Sanz, D.; Bruña, R.; Delgado-Losada, M.L.; López-Higes, R.; Marcos-Dolado, A.; Maestú, F.; Walter, S. Electrophysiological brain signatures for the classification of subjective cognitive decline: Towards an individual detection in the preclinical stages of dementia. Alzheimer’s Res. Ther. 2019, 11, 49.
- Sharma, V.K.; Singh, T.G.; Singh, S.; Garg, N.; Dhiman, S. Apoptotic Pathways and Alzheimer’s Disease: Probing Therapeutic Potential. Neurochem. Res. 2021, 46, 3103–3122.
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479.
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161.
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310.
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21.
- Leelakanok, N.; D’Cunha, R.R. Association between polypharmacy and dementia—A systematic review and metaanalysis. Aging Ment. Health 2019, 23, 932–941.
- Maher, R.L.; Hanlon, J.; Hajjar, E.R. Clinical consequences of polypharmacy in elderly. Expert Opin. Drug Saf. 2014, 13, 57–65.
- Chang, A.; Rosani, A.; Quick, J. Capsaicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Reyes-Escogido Mde, L.; Gonzalez-Mondragon, E.G.; Vazquez-Tzompantzi, E. Chemical and pharmacological aspects of capsaicin. Molecules 2011, 16, 1253–1270.
- O’Neill, J.; Brock, C.; Olesen, A.E.; Andresen, T.; Nilsson, M.; Dickenson, A.H. Unravelling the mystery of capsaicin: A tool to understand and treat pain. Pharmacol. Rev. 2012, 64, 939–971.
- Rollyson, W.D.; Stover, C.A.; Brown, K.C.; Perry, H.E.; Stevenson, C.D.; McNees, C.A.; Ball, J.G.; Valentovic, M.A.; Dasgupta, P. Bioavailability of capsaicin and its implications for drug delivery. J. Control. Release 2014, 196, 96–105.
- Govindarajan, V.S.; Sathyanarayana, M.N. Capsicum--production, technology, chemistry, and quality. Part V. Impact on physiology, pharmacology, nutrition, and metabolism; structure, pungency, pain, and desensitization sequences. Crit. Rev. Food Sci. Nutr. 1991, 29, 435–474.
- Fattori, V.; Hohmann, M.S.; Rossaneis, A.C.; Pinho-Ribeiro, F.A.; Verri, W.A. Capsaicin: Current Understanding of Its Mechanisms and Therapy of Pain and Other Pre-Clinical and Clinical Uses. Molecules 2016, 21, 844.
- Meghvansi, M.K.; Siddiqui, S.; Khan, M.H.; Gupta, V.K.; Vairale, M.G.; Gogoi, H.K.; Singh, L. Naga chilli: A potential source of capsaicinoids with broad-spectrum ethnopharmacological applications. J. Ethnopharmacol. 2010, 132, 1–14.
- Shang, K.; Amna, T.; Amina, M.; Al-Musayeib, N.M.; Al-Deyab, S.S.; Hwang, I. Influence of Capsaicin on Inflammatory Cytokines Induced by Lipopolysaccharide in Myoblast Cells Under In vitro Environment. Pharmacogn. Mag. 2017, 13, S26–S32.
- Tang, J.; Luo, K.; Li, Y.; Chen, Q.; Tang, D.; Wang, D.; Xiao, J. Capsaicin attenuates LPS-induced inflammatory cytokine production by upregulation of LXRα. Int. Immunopharmacol. 2015, 28, 264–269.
- Galano, A.; Martínez, A. Capsaicin, a tasty free radical scavenger: Mechanism of action and kinetics. J. Phys. Chem. B 2012, 116, 1200–1208.
- Lu, M.; Chen, C.; Lan, Y.; Xiao, J.; Li, R.; Huang, J.; Huang, Q.; Cao, Y.; Ho, C.T. Capsaicin-the major bioactive ingredient of chili peppers: Bio-efficacy and delivery systems. Food Funct. 2020, 11, 2848–2860.
- Abdel-Salam, O.M.E.; Sleem, A.A.; Sayed, M.; Youness, E.R.; Shaffie, N. Capsaicin Exerts Anti-convulsant and Neuroprotective Effects in Pentylenetetrazole-Induced Seizures. Neurochem. Res. 2020, 45, 1045–1061.
- Khatibi, N.H.; Jadhav, V.; Charles, S.; Chiu, J.; Buchholz, J.; Tang, J.; Zhang, J.H. Capsaicin pre-treatment provides neurovascular protection against neonatal hypoxic-ischemic brain injury in rats. Acta Neurochir. Suppl. 2011, 111, 225–230.
- Xu, W.; Liu, J.; Ma, D.; Yuan, G.; Lu, Y.; Yang, Y. Capsaicin reduces Alzheimer-associated tau changes in the hippocampus of type 2 diabetes rats. PLoS ONE 2017, 12, e0172477.
- Liu, C.H.; Bu, X.L.; Wang, J.; Zhang, T.; Xiang, Y.; Shen, L.L.; Wang, Q.H.; Deng, B.; Wang, X.; Zhu, C.; et al. The Associations between a Capsaicin-Rich Diet and Blood Amyloid-β Levels and Cognitive Function. J. Alzheimer’s Dis. 2016, 52, 1081–1088.
- Tian, D.Y.; Wang, J.; Sun, B.L.; Wang, Z.; Xu, W.; Chen, Y.; Shen, Y.Y.; Li, H.Y.; Chen, D.W.; Zhou, F.Y.; et al. Spicy food consumption is associated with cognition and cerebrospinal fluid biomarkers of Alzheimer disease. Chin. Med. J. 2020, 134, 173–177.
- Shi, Z.; El-Obeid, T.; Riley, M.; Li, M.; Page, A.; Liu, J. High Chili Intake and Cognitive Function among 4582 Adults: An Open Cohort Study over 15 Years. Nutrients 2019, 11, 1183.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
03 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No