Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oliver Moore | -- | 2550 | 2023-06-29 20:45:42 | | | |
| 2 | Sirius Huang | Meta information modification | 2550 | 2023-07-03 02:37:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Moore, O.M.; Ho, K.S.; Copeland, J.S.; Parthasarathy, V.; Wehrens, X.H.T. Genome Editing in Treating Cardiac Arrhythmias. Encyclopedia. Available online: https://encyclopedia.pub/entry/46240 (accessed on 24 July 2026).
Moore OM, Ho KS, Copeland JS, Parthasarathy V, Wehrens XHT. Genome Editing in Treating Cardiac Arrhythmias. Encyclopedia. Available at: https://encyclopedia.pub/entry/46240. Accessed July 24, 2026.
Moore, Oliver M., Kevin S. Ho, Juwan S. Copeland, Vaidya Parthasarathy, Xander H. T. Wehrens. "Genome Editing in Treating Cardiac Arrhythmias" Encyclopedia, https://encyclopedia.pub/entry/46240 (accessed July 24, 2026).
Moore, O.M., Ho, K.S., Copeland, J.S., Parthasarathy, V., & Wehrens, X.H.T. (2023, June 29). Genome Editing in Treating Cardiac Arrhythmias. In Encyclopedia. https://encyclopedia.pub/entry/46240
Moore, Oliver M., et al. "Genome Editing in Treating Cardiac Arrhythmias." Encyclopedia. Web. 29 June, 2023.
Copy Citation
Despite advances in screening and preventative treatments, the estimated lifetime risk for premature death due to arrhythmogenic sudden cardiac death remains high. A growing understanding of the genetics underlying cardiac arrhythmias has enabled new treatment possibilities including the use of cardiac genome editing.
arrhythmias
catecholaminergic polymorphic ventricular tachycardia
CRISPR/Cas9
genome editing
1. Genome Editing Techniques
While gene targeting in embryonic cells has been used for modeling familial cardiac disorders, somatic cardiac genome editing only became feasible following the development of nucleases that can introduce double-stranded breaks (DSBs) at specific locations in the genome. These nucleases can be categorized into four general categories: meganucleases, zinc-finger nucleases (ZFN), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR) associated with nuclease 9 (Cas9) [1]. Notably, however, only the CRISPR/Cas9 system has been used for therapeutic somatic genome editing of the heart.
In the CRISPR/Cas9 system, a synthetic RNA spacer sequence is inserted into a scaffold for the Cas9 nuclease [2][3]. This guide RNA (gRNA) sequence allows Cas9 to target genomic sequences of 20 or more base pairs (bp). Cas9 binds DNA at the protospacer adjacent motif (PAM) and cleaves 3 bp upstream of the PAM (Figure 1). In contrast to the amino acid recognition sequences that other endonucleases rely on, the CRISPR/Cas9 spacer sequences are much simpler to design, synthesize, and validate. As such, the field of genome editing overall has largely moved to this platform, and subsequent discoveries in cardiac gene editing have relied on utilizing the CRISPR/Cas9 system. The first publication of somatic genome editing of the heart involved using this system to correct Duchenne’s muscular dystrophy (DMD) in a mouse model [4]. Subsequently, there have been several developments in expanding the capabilities and efficacy of the CRISPR/Cas9 systems and these are reviewed in the subsequent sections [5].
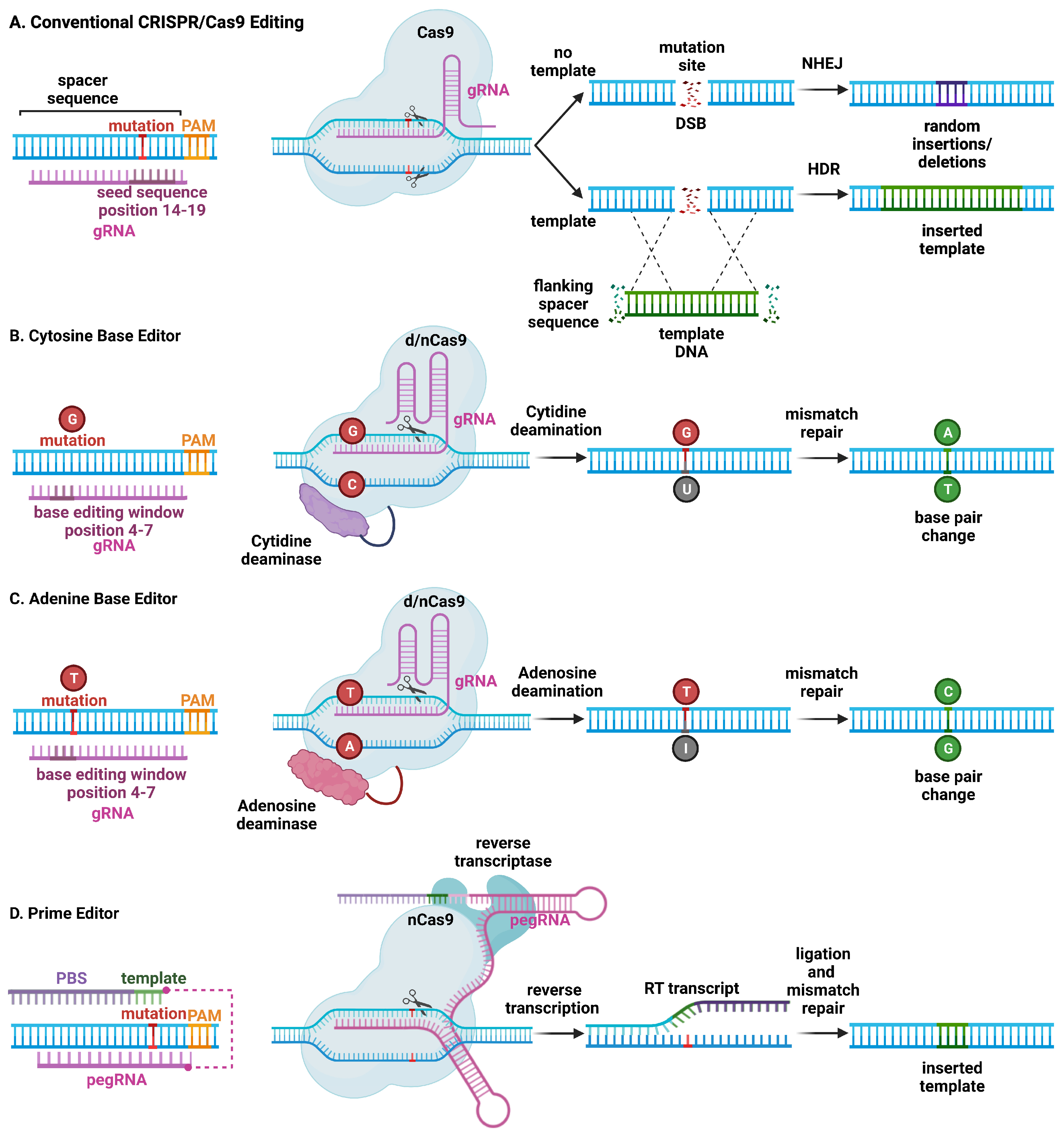
Figure 1. Schematic overview of major CRISPR/Cas9-based genome editing technologies. (A) Conventional CRISPR/Cas9 editing of a mutant allele involves selecting guide RNAs adjacent to a protospacer adjacent motif (PAM) site, with a mutation in the spacer sequence (14–19) to minimize wild-type allele cleavage. The Cas9 endonuclease introduces a double-stranded break (DSB). Non-homologous end joining (NHEJ) repairs the mutant allele but introduces insertions and deletions at the mutation site, which can lead to frame-shift mutations and stop codons, resulting in nonsense-mediated decay of mutant RNA (top). On the other hand, homology-directed repair (HDR) is less efficient but leads to correction of the mutant allele using a DNA repair template. (B) Cytosine base editors are created by fusing Cas9 nickase (nCas9) or catalytically inactive “dead” Cas9 (dCas9) to a cytidine deaminase. Base editors are targeted to a specific locus using gRNA. They can convert cytidine (C) to uridine (U) within a small editing window (4–7 in the spacer sequence, depending on the editor type), near the PAM site. Uridine is subsequently converted to thymidine (T) through base excision repair. (C) Likewise, adenosine base editors have been engineered to convert adenosine (A) to inosine (I), which is subsequently converted to guanidine (G). (D) Prime editing involves a reverse transcriptase and dCas9 that can produce single-stranded DNA breaks. The 3′-extended single guide RNA contains a primer binding site and a reverse transcriptase template, which is referred to as primer editing guide RNA (pegRNA). Hybridization of the exposed 3′-end to the primer binding site primes the reverse transcription of the nicked DNA strand for the desired edit.
The expression of CRISPR/Cas9 with a single gRNA is sufficient to create a DSB at a target genome locus. In cardiomyocytes, this has been shown to cause primarily non-homologous end-joining (NHEJ) repair (Figure 1) [6]. The lack of fidelity in NHEJ repair, however, results in insertions and deletions (indels) that can result in a frame shift, which—when introduced within a coding sequence—may lead to nonsense-mediated degradation. This method has been employed to model loss-of-function mutations by disrupting a target gene. In addition, this method can be used to disable a toxic or detrimental gain-of-function allele in heterozygous individuals. The feasibility of postnatal gene editing was demonstrated in mice overexpressing a cardiomyocyte-specific Cas9 in combination with adeno-associated viral (AAV) vector-mediated delivery of guide RNAs targeting Myh6, Sav1, and Tbx20 [7]. Following AAV delivery, mosaic disruption of the target genes was seen, and only disruption of Myh6 was sufficient to induce a cardiac phenotype. Despite the limitations of a mosaic knockdown in terms of efficiency, this method has been uniquely beneficial for characterizing a novel loss-of-function arrhythmia disorder that would otherwise be lethal in a complete knockout state [8]. NHEJ editing has also been effective in deleting splice donor or acceptor site sequences in out-of-frame exons, thereby restoring the open reading frame of the gene of interest. Several groups have demonstrated the restoration of cardiac dystrophin levels using AAV-mediated delivery of Cas9 and a single gRNA in mouse models of Duchenne muscular dystrophy DMD [9][10][11][12].
CRISPR/Cas9 genome editing can also be used to replace and correct the target sequence with a template through homology-directed repair (HDR) (Figure 1). Although gene correction is attained, HDR is less efficient compared with NHEJ, and a vast majority of edits will contain random insertions or deletions. Given the low efficiency in non-dividing cells such as cardiomyocytes, HDR has not been used (yet) for the treatment of cardiac arrhythmias in preclinical animal models. Another CRISPR technique involves using at least one gRNA sequence and a template sequence flanked by the gRNA target sequences in reverse orientation to create a DSB at the target site, followed by insertion of a corrected gene by incorporation of the template DNA through homology-independent targeted integration (HITI) [13]. Due to the limited efficacy of HDR in post mitotic tissue, HITI has not been used to correct arrhythmia phenotypes, although it was used to increase full-length dystrophin expression in a humanized mouse model of DMD with a genomic correction rate of 4–7% in cardiomyocytes [14].
Alternative CRISPR/Cas9-based genome editing techniques have been developed that allow for the introduction of point mutations in the genomic DNA without generating DSBs [15]. By fusing Cas9 nickase (nCas9) or deactivated Cas9 (dCas9) to a deaminase enzyme, the resulting base editor can edit DNA without DSBs, converting C to T, or G to A. Further development of cytosine base editors (CBE) and adenine base editors (ABE) resulted in expansions of possible conversions including C to T, A to G, T to C, and G to A, respectively [15]. Studies revealed an in vitro efficiency of 15–75%, with less than 1% indel formation, although efficiency is much lower in non-dividing cells. Moreover, DNA base editors have some shortcomings, including off-target DNA editing, the generation of bystander mutations, and promiscuous deamination effects in both DNA and RNA. Finally, prime editing uses a fusion protein, consisting of a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase, and a prime editing guide RNA (pegRNA), capable of replacing the target DNA nucleotides without the need for DSBs or donor DNA templates [16].
2. Using CRISPR/Cas9 Genome Editing to Create Animal and Cellular Arrhythmia Models
The advent of genome editing approaches such as CRISPR/Cas9 has created a whole new set of possibilities to generate animal models for biomedical research using virtually any species. Animal models in which human gene variants are introduced can provide convincing evidence for a disease-causing pathogenic mechanism [17]. While gene overexpression or knockdown strategies may provide useful insights in some cases [18], knock-in strategies that involve introducing the exact genetic variant in the model system are generally superior [19]. The RNA-guided Cas9 nuclease system can be used to easily generate knock-in mouse models [20]. This system has superseded all other systems for genome editing because of its simplicity of use, lower costs, and higher efficiency [21]. These days, most researchers prefer this approach over ES cell-based gene-targeting methods [22]. The CRISPR/Cas9 system allows for the introduction of footprint-free point mutations on various genetic backgrounds. While many of the initial mouse models of monogenetic arrhythmia syndromes were generated using ES cell-based methods, several newer models have been generated using CRISPR/Cas9 gene editing [23][24].
Genome editing can also be used to introduce potential disease-causing variants in patient-derived induced pluripotent stem cells (iPSCs) that can subsequently be differentiated into cardiomyocytes [25]. This approach has been particularly helpful to elucidate whether genetic variants of unknown significance (VUSs) in various genes purported to cause arrhythmias are in fact pathogenic [26]. Various genome editing techniques including CRISPR/Cas9 can introduce or correct genetic variants in iPSC before differentiation into cardiomyocytes, providing high throughput empirical evidence for characterizing novel mutations, their mechanisms, and potential therapies [27]. Many iPSC models of heritable arrhythmias, including LQTS, Brugada syndrome, CPVT, and arrhythmogenic cardiomyopathy, have been generated, as recently reviewed by Yang et al. [28]. These models have been used not only to characterize disease mechanisms but also to test the effectiveness of novel therapeutics [29].
In addition to being a tool for introducing genetic variants into iPSCs, CRISPR/Cas9 also enables efficient correction of inherited variants in iPSCs. By taking somatic cells such as skin fibroblasts or peripheral whole blood samples from patients, patient-derived iPSCs can be generated that include all VUSs present in the patient genome. Generating an isogenic control provides incredibly powerful evidence on whether a specific SNP is likely to be pathogenic. Most studies to this effect have used HDR to generate isogenic controls [30]. However, recent studies have begun to use more advanced techniques such as base editing and prime editing to correct specific variants in iPSCs [31]. The ease with which iPSCs can undergo genome editing has dramatically increased the number of VUS that can be characterized and has also expanded the genetic approaches that can be used to explore disease mechanisms. There are important limitations to the use of iPSC-derived cardiomyocytes (iPSC-CMs). While studies in single iPSC-CMs might yield new insights about certain inherited arrhythmia syndromes, it would be implausible that relevant insights could be gleaned from iPSC-CMs generated from patients with atrial fibrillation or ischemic heart disease. In addition, despite improvements in experimental protocols, iPSC-CMs still exhibit relatively immature and variable phenotypes. Finally, iPSC-CMs often do not have a functional SR, and the intracellular Ca2+ handling dynamics are quite different from those in freshly isolated CMs from adult animals or human patients [32].
3. Therapeutic Genome Editing in Preclinical Arrhythmia Models
Somatic genome editing has also been used to correct heart disease in preclinical model systems (Figure 2). For example, the CRISPR/Cas9 editing method was employed in correcting the cardiac PRKAG2 syndrome, which is known to cause familial Wolff–Parkinson–White syndrome with cardiomyopathy [33]. PRKAG2 syndrome is an autosomal-dominant inherited disease caused by missense mutations in the PRKAG2 gene, which encodes the γ2 regulatory subunit of AMP-activated protein kinase [34]. Altered activity of this AMP-activated protein kinase results in excessive cellular glycogen deposition leading to cardiomyopathy and supraventricular arrhythmias. Systemic administration of adeno-associated virus serotype 9 (AAV9) with gRNA and the CRISPR/spCas9 gene-editing system was sufficient to disrupt the mutant PRKAG2 allele in mice while leaving the wild-type allele intact [35]. While the genome-editing efficiency was relatively low (∼6.5% in mice injected at postnatal day (P) 4 and ∼2.6% in mice injected at P42), this treatment strategy reduced preexcitation arrhythmias by 40%, and restored the morphology and function of the heart in mutant mice [35].
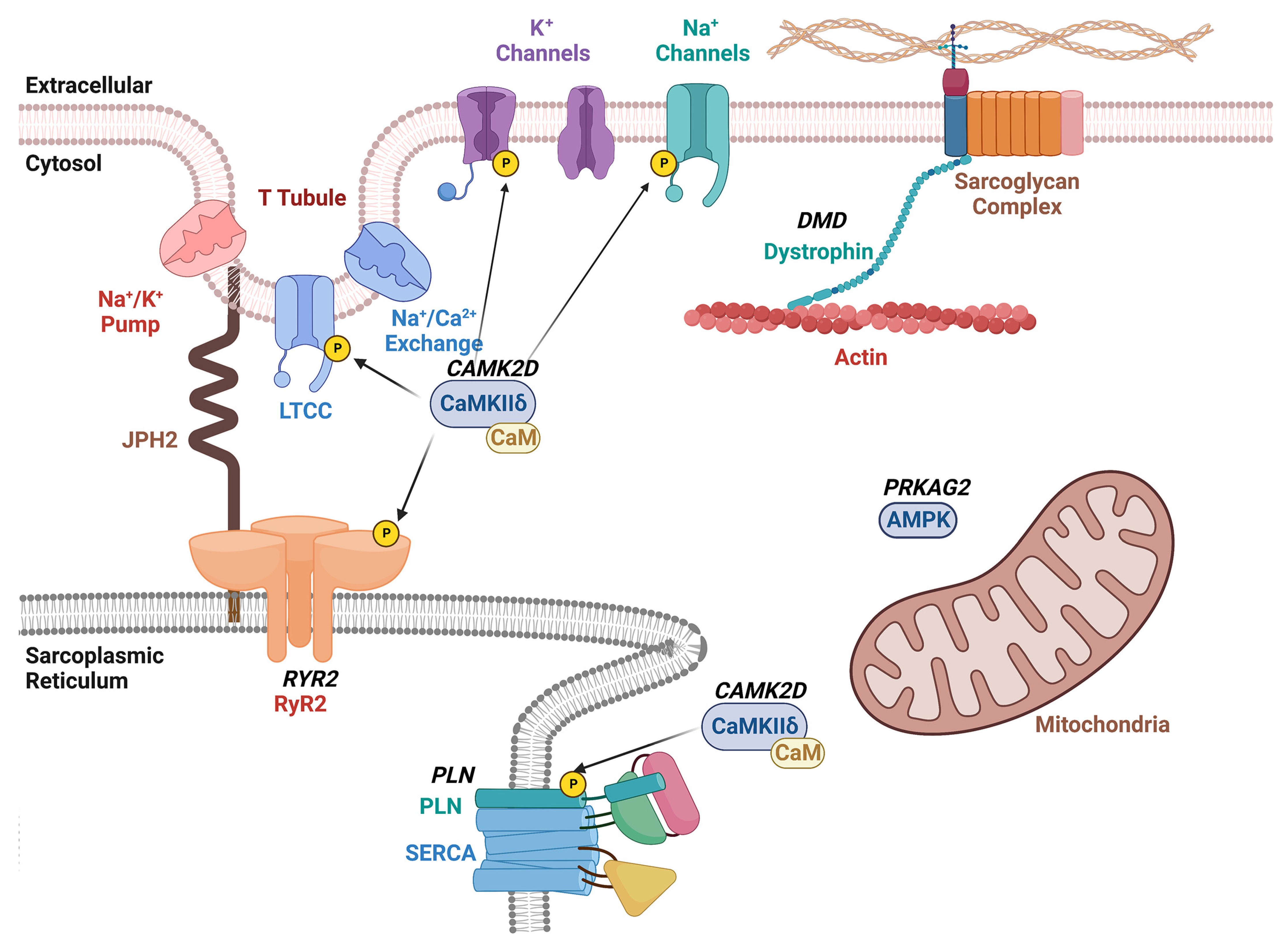
Figure 2. Schematic overview of therapeutic CRISPR/Cas9 targets for arrhythmia treatment. The diagram shows the plasma membrane with various ion channels including the voltage-gated L-type Ca2+ channel (LTCC), and K+ and Na+ channels. The sarcoplasmic reticulum (SR) is shown with the ryanodine receptor type-2 (RyR2)/Ca2+ release channel and sarco-/endoplasmic Ca2+-ATPase (SERCA2a) with its regulatory subunit phospholamban (PLN). Dystrophin links the plasmalemmal sarcoglycan complex to the sarcomere. The Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylates various ion channels and Ca2+ handling proteins within cardiomyocytes. Gene names that are targets of therapeutic genome editing are indicated in bold and italicized.
One research group utilized a similar approach to treat catecholaminergic polymorphic ventricular tachycardia (CPVT) in a preclinical mouse model [36]. CPVT is an autosomal-dominant inherited disease caused primarily by missense mutations in the RYR2 gene that encodes the ryanodine receptor type 2 (RyR2) intracellular calcium channel [37]. The mutated channel generates a diastolic calcium leak that can trigger lethal arrhythmias [19]. Researchers found that systemic administration of AAV to deliver gRNA and SaCas9 was sufficient to disrupt the mutant RyR2 allele in a heterozygous mice model while leaving the wild-type allele intact. The editing frequency based on next generation sequencing was found to be around 11% in these RyR2 mutant mice [36]. In addition, the reduced mutant allele mRNA levels were indicative of nonsense-mediated decay. This degree of genome editing reduced the ventricular tachycardia incidence by 100% and normalized channel function at the cellular level [36].
The same NHEJ pathway was induced by CRISPR/Cas9 genome editing in a humanized mouse model of arrhythmogenic dilated cardiomyopathy caused by a truncating variant in phospholamban (PLN) [38]. The PLN-R14del mutation was first discovered in a large Greek family with clinical signs of both dilated cardiomyopathy (DCM) and arrhythmogenic cardiomyopathy (ACM) [39]. Subsequently, many PLN-R14del mutation carriers were found in the Netherlands, where the original mutation is believed to have originated [40]. Mice harboring the R14del mutant human PLN were found to be more susceptible to rapid pacing-induced ventricular tachycardia. In vivo gene editing using AAV9 led to a greater than two-fold reduction in pacing-induced VT incidence and significantly raised the frequency threshold for induction [38].
While gene disruption of dominant-negative mutations is one of the most efficient methods of therapy, other methods of genome editing are needed in the case of haplo-insufficiency. Duchenne muscular dystrophy (DMD) is a fatal X-linked recessive disorder that causes progressive neuromuscular weakness and wasting. A quarter of DMD patients die from cardiac causes, and half of these deaths are due to lethal ventricular tachycardia [41]. Abnormal Ca2+ release from the sarcoplasmic reticulum via RyR2 overactivated by Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a major mechanism underlying arrhythmogenesis in mice with DMD [42]. Inherited variants in the DMD gene usually involve single or multi-exon deletions that disrupt the open reading frame (ORF) and introduce a premature stop codon, resulting in a nonfunctional, truncate dystrophin protein [43]. CRISPR/Cas9 has been used to generate indels using NHEJ in the DMD gene to restore the ORF in rodent and large animal preclinical models [9]. Recently, Chemello et al. [31] used an adenine base editor (ABE) delivered using AAV9 as a split-intein system to restore protein expression in a DMD mouse model. By means of a single-swap base pair transition in the dinucleotide splicing motif, exon skipping was accomplished with restoration of dystrophin levels. The on-target editing efficiency ranged from 6.7 to 35.0%, with no notable editing at off-target sites [31]. Whereas the effects of base editing on arrhythmogenesis were not assessed in vivo, complementary studies in human induced-pluripotent stem cell-derived cardiomyocytes revealed that base editing normalized arrhythmic calcium handling deficits [31]. Additional studies are needed to determine whether base editing can also block lethal arrhythmias in vivo in animal models of DMD.
Finally, it may be possible to use genome editing for the treatment on non-genetic arrhythmia disorders by targeting key molecular switches that drive disease development. For example, Lebek et al. [44] recently showed that base editing could be used to eliminate oxidation-sensitive methionine residues on CaMKII [45] to confer protection from ischemia/reperfusion injury, which is often associated with ventricular arrhythmias. Since the same CaMKII residues have been implicated in ventricular arrhythmogenesis in DMD [42] and atrial fibrillation [46], similar approaches may hold promise for a wider array of arrhythmia disorders.
References
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403.
- Roshanravan, N.; Tutunchi, H.; Najafipour, F.; Dastouri, M.; Ghaffari, S.; Jebeli, A. A glance at the application of CRISPR/Cas9 gene-editing technology in cardiovascular diseases. J. Cardiovasc. Thorac. Res. 2022, 14, 77–83.
- Vermersch, E.; Jouve, C.; Hulot, J.S. CRISPR/Cas9 gene-editing strategies in cardiovascular cells. Cardiovasc. Res. 2020, 116, 894–907.
- Johansen, A.K.; Molenaar, B.; Versteeg, D.; Leitoguinho, A.R.; Demkes, C.; Spanjaard, B.; de Ruiter, H.; Akbari Moqadam, F.; Kooijman, L.; Zentilin, L.; et al. Postnatal Cardiac Gene Editing Using CRISPR/Cas9 with AAV9-Mediated Delivery of Short Guide RNAs Results in Mosaic Gene Disruption. Circ. Res. 2017, 121, 1168–1181.
- Guo, Y.; Cao, Y.; Jardin, B.D.; Zhang, X.; Zhou, P.; Guatimosim, S.; Lin, J.; Chen, Z.; Zhang, Y.; Mazumdar, N.; et al. Ryanodine receptor 2 (RYR2) dysfunction activates the unfolded protein response and perturbs cardiomyocyte maturation. Cardiovasc. Res. 2023, 119, 221–235.
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, 418.
- Zhang, Y.; Li, H.; Min, Y.L.; Sanchez-Ortiz, E.; Huang, J.; Mireault, A.A.; Shelton, J.M.; Kim, J.; Mammen, P.P.A.; Bassel-Duby, R.; et al. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci. Adv. 2020, 6, eaay6812.
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91.
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324.
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149.
- Pickar-Oliver, A.; Gough, V.; Bohning, J.D.; Liu, S.; Robinson-Hamm, J.N.; Daniels, H.; Majoros, W.H.; Devlin, G.; Asokan, A.; Gersbach, C.A. Full-length dystrophin restoration via targeted exon integration by AAV-CRISPR in a humanized mouse model of Duchenne muscular dystrophy. Mol. Ther. 2021, 29, 3243–3257.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993.
- Beavers, D.L.; Wang, W.; Ather, S.; Voigt, N.; Garbino, A.; Dixit, S.S.; Landstrom, A.P.; Li, N.; Wang, Q.; Olivotto, I.; et al. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J. Am. Coll. Cardiol. 2013, 62, 2010–2019.
- Kannankeril, P.J.; Mitchell, B.M.; Goonasekera, S.A.; Chelu, M.G.; Zhang, W.; Sood, S.; Kearney, D.L.; Danila, C.I.; De Biasi, M.; Wehrens, X.H.; et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 12179–12184.
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723.
- Gurumurthy, C.B.; Lloyd, K.C.K. Generating mouse models for biomedical research: Technological advances. Dis. Model Mech. 2019, 12, dmm029462.
- Gordon, J.W.; Ruddle, F.H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 1981, 214, 1244–1246.
- Tsai, W.C.; Guo, S.; Olaopa, M.A.; Field, L.J.; Yang, J.; Shen, C.; Chang, C.P.; Chen, P.S.; Rubart, M. Complex Arrhythmia Syndrome in a Knock-In Mouse Model Carrier of the N98S Calm1 Mutation. Circulation 2020, 142, 1937–1955.
- Lubberding, A.F.; Zhang, J.; Lundh, M.; Nielsen, T.S.; Sondergaard, M.S.; Villadsen, M.; Skovhoj, E.Z.; Boer, G.A.; Hansen, J.B.; Thomsen, M.B.; et al. Age-dependent transition from islet insulin hypersecretion to hyposecretion in mice with the long QT-syndrome loss-of-function mutation Kcnq1-A340V. Sci. Rep. 2021, 11, 12253.
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857.
- van Lint, F.H.M.; Mook, O.R.F.; Alders, M.; Bikker, H.; Lekanne Dit Deprez, R.H.; Christiaans, I. Large next-generation sequencing gene panels in genetic heart disease: Yield of pathogenic variants and variants of unknown significance. Neth. Heart J. 2019, 27, 304–309.
- Yoshinaga, D.; Baba, S.; Makiyama, T.; Shibata, H.; Hirata, T.; Akagi, K.; Matsuda, K.; Kohjitani, H.; Wuriyanghai, Y.; Umeda, K.; et al. Phenotype-Based High-Throughput Classification of Long QT Syndrome Subtypes Using Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 394–404.
- Yang, B.; Lowenthal, J.; Tomaselli, G.F.; Tung, L. Human iPSC models of cardiac electrophysiology and arrhythmia. In iPSCs—State of the Science; Birbrair, A., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 16, pp. 29–93.
- Song, Y.; Guo, T.; Jiang, Y.; Zhu, M.; Wang, H.; Lu, W.; Jiang, M.; Qi, M.; Lan, F.; Cui, M. KCNQ1-deficient and KCNQ1-mutant human embryonic stem cell-derived cardiomyocytes for modeling QT prolongation. Stem Cell Res. Ther. 2022, 13, 287.
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096.
- Chemello, F.; Chai, A.C.; Li, H.; Rodriguez-Caycedo, C.; Sanchez-Ortiz, E.; Atmanli, A.; Mireault, A.A.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv. 2021, 7, 18.
- Word, T.A.; Quick, A.P.; Miyake, C.Y.; Shak, M.K.; Pan, X.; Kim, J.J.; Allen, H.D.; Sibrian-Vazquez, M.; Strongin, R.M.; Landstrom, A.P.; et al. Efficacy of RyR2 inhibitor EL20 in induced pluripotent stem cell-derived cardiomyocytes from a patient with catecholaminergic polymorphic ventricular tachycardia. J. Cell Mol. Med. 2021, 25, 6115–6124.
- Aggarwal, V.; Dobrolet, N.; Fishberger, S.; Zablah, J.; Jayakar, P.; Ammous, Z. PRKAG2 mutation: An easily missed cardiac specific non-lysosomal glycogenosis. Ann. Pediatr. Cardiol. 2015, 8, 153–156.
- Wolf, C.M.; Arad, M.; Ahmad, F.; Sanbe, A.; Bernstein, S.A.; Toka, O.; Konno, T.; Morley, G.; Robbins, J.; Seidman, J.G.; et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation 2008, 117, 144–154.
- Xie, C.; Zhang, Y.P.; Song, L.; Luo, J.; Qi, W.; Hu, J.; Lu, D.; Yang, Z.; Zhang, J.; Xiao, J.; et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016, 26, 1099–1111.
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963.
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840.
- Dave, J.; Raad, N.; Mittal, N.; Zhang, L.; Fargnoli, A.; Oh, J.G.; Savoia, M.E.; Hansen, J.; Fava, M.; Yin, X.; et al. Gene editing reverses arrhythmia susceptibility in humanized PLN-R14del mice: Modelling a European cardiomyopathy with global impact. Cardiovasc. Res. 2022, 118, 3140–3150.
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393.
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207.
- Ather, S.; Wang, W.; Wang, Q.; Li, N.; Anderson, M.E.; Wehrens, X.H. Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm 2013, 10, 592–599.
- Wang, Q.; Quick, A.P.; Cao, S.; Reynolds, J.; Chiang, D.Y.; Beavers, D.; Li, N.; Wang, G.; Rodney, G.G.; Anderson, M.E.; et al. Oxidized CaMKII (Ca(2+)/Calmodulin-Dependent Protein Kinase II) Is Essential for Ventricular Arrhythmia in a Mouse Model of Duchenne Muscular Dystrophy. Circ. Arrhythm. Electrophysiol. 2018, 11, e005682.
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740.
- Lebek, S.; Chemello, F.; Caravia, X.M.; Tan, W.; Li, H.; Chen, K.; Xu, L.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Ablation of CaMKIIdelta oxidation by CRISPR-Cas9 base editing as a therapy for cardiac disease. Science 2023, 379, 179–185.
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474.
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757.
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
587
Revisions:
2 times
(View History)
Update Date:
03 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No