 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ya-Zhou Shi | -- | 2098 | 2023-06-27 12:14:14 | | | |
| 2 | Jessie Wu | + 4 word(s) | 2102 | 2023-06-28 04:44:04 | | | | |
| 3 | Jessie Wu | -3 word(s) | 2099 | 2023-06-28 04:45:10 | | |
Video Upload Options
DNA carries the genetic information required for the synthesis of RNA and proteins and plays an important role in many processes of biological development. Understanding the three-dimensional (3D) structures and dynamics of DNA is crucial for understanding their biological functions and guiding the development of novel materials. Molecular dynamics (MD) simulations can generally reproduce the behavior of DNAs in a computer, providing detailed structural and dynamical insights that enhancing our comprehension of relevant experimental data. MD simulations using classical force fields such as AMBER and CHARMM have provided highly detailed and flexible descriptions of DNA dynamics, including structural transformations, stability of non-canonical conformations, salt ion cohesion effects, twist-stretch coupling of stress, flexibility under methylation modifications, and interactions with other macromolecules. It is always fascinating to obtain microscopic insights into DNA dynamics through MD simulations. However, the innumerable degrees of freedom, interconnected in complex ways, can make it practically impossible to detect DNA dynamics on biologically relevant time scales and length scales using currently available computer hardware
1. Introduction
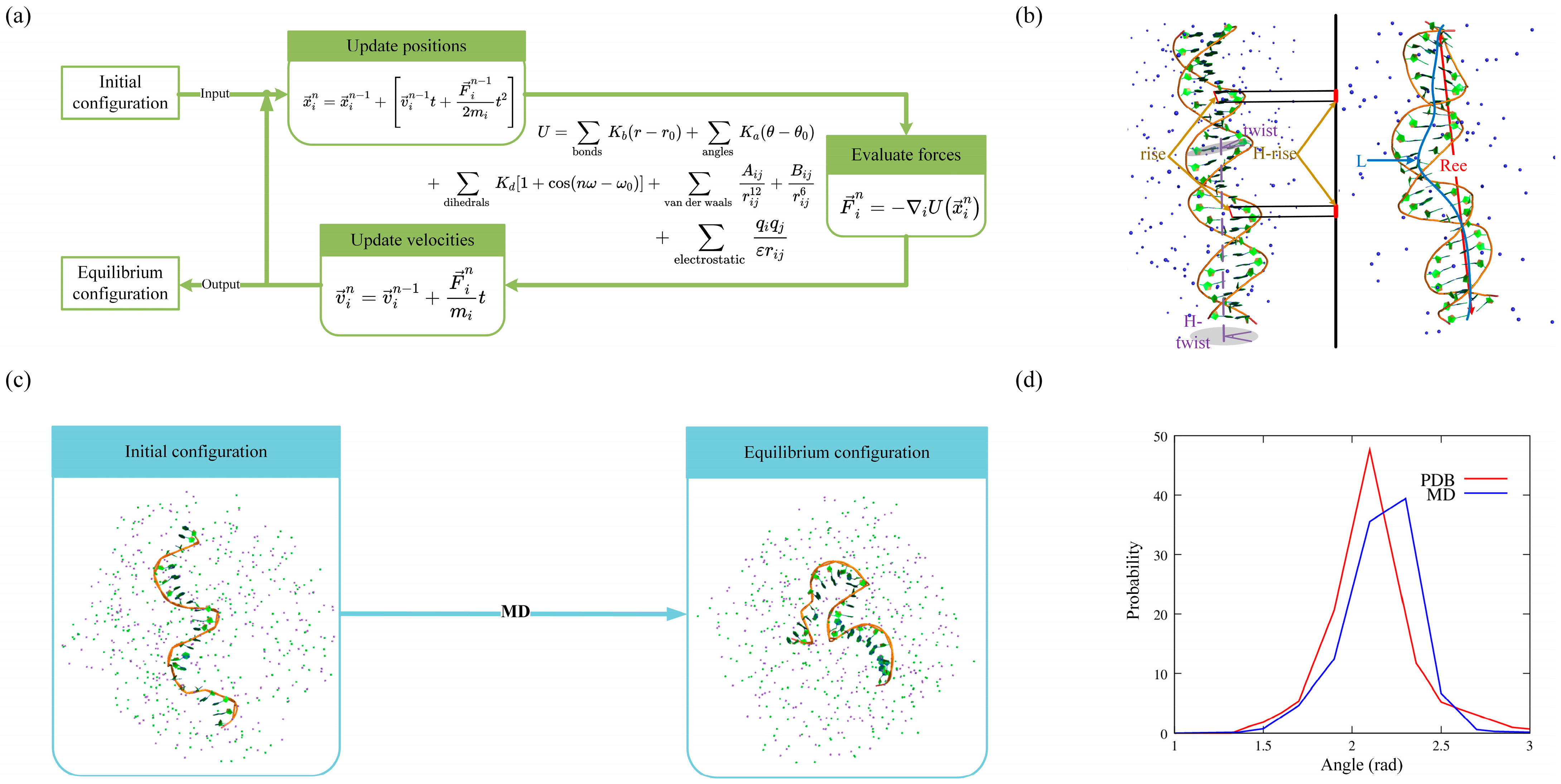
2. Structural Dynamics
3. Structural Flexibility
4. DNA–Ion Interaction
5. Limitations
References
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2005, 26, 114.
- Zhang, Y.; Zhang, Y.; McCready, M.J.; Maginn, E.J. Evaluation and refinement of the general AMBER force field for nineteen pure organic electrolyte solvents. J. Chem. Eng. 2018, 3488–3502.
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; MacKerell, A.D., Jr. Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362.
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Kruger, P.; Van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. 1999, 103, 3596–3607.
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509.
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58.
- Chen, A.A.; García, A.E. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simula-tions. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825.
- Gallardo, A.; Bogart, B.M.; Dutagaci, B. Protein–Nucleic Acid Interactions for RNA Polymerase II Elongation Factors by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 3079–3089.
- Salsbury, A.M.; A Lemkul, J. Recent developments in empirical atomistic force fields for nucleic acids and applications to studies of folding and dynamics. Curr. Opin. Struct. Biol. 2021, 67, 9–17.
- Kameda, T.; Awazu, A.; Togashi, Y. Molecular dynamics analysis of biomolecular systems including nucleic acids. Biophys. Physicobiology 2022, 19, e190027.
- Ghoshdastidar, D.; Bansal, M. Dynamics of physiologically relevant noncanonical DNA structures: An overview from experi-mental and theoretical studies. Brief. Funct. Genom. 2018, 18, 192–204.
- Frank-Kamenetskii, M.D.; Prakash, S. Fluctuations in the DNA double helix: A critical review. Phys. Life Rev. 2014, 11, 153–170.
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736.
- Strelnikov, I.A.; Kovaleva, N.A.; Klinov, A.P.; Zubova, E.A. C-B-A test of DNA force fields. ACS Omega 2023, 8, 10253–10265.
- Panczyk, T.; Wojton, P.; Wolski, P. Mechanism of unfolding and relative stabilities of G-quadruplex and I-motif noncanonical DNA structures analyzed in biased molecular dynamics simulations. Biophys. Chem. 2019, 250, 106173.
- Panczyk, T.; Wojton, P.; Wolski, P. Molecular Dynamics Study of the Interaction of Carbon Nanotubes with Telomeric DNA Fragment Containing Noncanonical G-Quadruplex and i-Motif Forms. Int. J. Mol. Sci. 2020, 21, 1925.
- Liu, T.; Yu, T.; Zhang, S.; Wang, Y.; Zhang, W. Thermodynamic and kinetic properties of a single base pair in A-DNA and B-DNA. Phys. Rev. E 2021, 103, 042409.
- Xu, S.; Zhan, J.; Man, B.; Jiang, S.; Yue, W.; Gao, S.; Guo, C.; Liu, H.; Li, Z.; Wang, J.; et al. Real-time reliable determination of binding kinetics of DNA hybridization using a multi-channel graphene biosensor. Nat. Commun. 2017, 8, 14902.
- Galindo-Murillo, R.; Roe, D.R. Cheatham TE 3rd. On the absence of intrahelical DNA dynamics on the μs to ms timescale. Nat. Commun. 2014, 5, 5152.
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta 2015, 1850, 1041–1058.
- Nikolova, E.N.; Kim, E.; Wise, A.A.; O’brien, P.J.; Andricioaei, I.; Al-Hashimi, H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 2011, 470, 498–502.
- Yang, C.; Kim, E.; Pak, Y. Free energy landscape and transition pathways from Watson–Crick to Hoogsteen base pairing in free duplex DNA. Nucleic Acids Res. 2015, 43, 7769–7778.
- Chakraborty, D.; Wales, D.J. Probing helical transitions in a DNA duplex. Phys. Chem. Chem. Phys. 2017, 19, 878.
- Marin-Gonzalez, A.; Vilhena, J.G.; Perez, R.; Moreno-Herrero, F. A molecular view of DNA flexibility. Q. Rev. Biophys. 2021, 54, e8.
- Ghoshdastidar, D.; Bansal, M. Flexibility of flanking DNA is a key determinant of transcription factor affinity for the core motif. Biophys. J. 2022, 121, 3987–4000.
- Nomidis, S.K.; Kriegel, F.; Vanderlinden, W.; Lipfert, J.; Carlon, E. Twist-Bend Coupling and the Torsional Response of Double-Stranded DNA. Phys. Rev. Lett. 2017, 118, 217801.
- Marko, J.F.; Siggia, E.D. Fluctuations and Supercoiling of DNA. Science 1994, 265, 506–508.
- Toan, N.M.; Thirumalai, D. On the origin of the unusual behavior in the stretching of single-stranded DNA. J. Chem. Phys. 2012, 136, 235103.
- Qiang, X.-W.; Dong, H.-L.; Xiong, K.-X.; Zhang, W.; Tan, Z.-J. Understanding sequence effect in DNA bending elasticity by molecular dynamic simulations. Commun. Theor. Phys. 2021, 73, 075601.
- Liebl, K.; Dršata, T.; Lankas, F.; Lipfert, J.; Zacharias, M. Explaining the striking difference in twist-stretch coupling between DNA and RNA: A comparative molecular dynamics analysis. Nucleic Acids Res. 2015, 43, 10143–10156.
- Chen, C.; Pettitt, B.M. The Effects of Flexibility on dsDNA-dsDNA Interactions. Life 2022, 12, 699.
- Wu, Y.-Y.; Bao, L.; Zhang, X.; Tan, Z.-J. Flexibility of short DNA helices with finite-length effect: From base pairs to tens of base pairs. J. Chem. Phys. 2015, 142, 125103.
- Marin-Gonzalez, A.; Vilhena, J.G.; Moreno-Herrero, F.; Perez, R. DNA Crookedness Regulates DNA Mechanical Properties at Short Length Scales. Phys. Rev. Lett. 2019, 122, 048102.
- Liebl, K.; Zacharias, M. Accurate modeling of DNA conformational flexibility by a multivariate Ising model. Proc. Natl. Acad. Sci. USA 2021, 118, e2021263118.
- Marin-Gonzalez, A.; Vilhena, J.G.; Perez, R.; Moreno-Herrero, F. Understanding the mechanical response of double-stranded DNA and RNA under constant stretching forces using all-atom molecular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, 7049–7054.
- Bao, L.; Zhang, X.; Shi, Y.-Z.; Wu, Y.-Y.; Tan, Z.-J. Understanding the Relative Flexibility of RNA and DNA Duplexes: Stretching and Twist-Stretch Coupling. Biophys. J. 2017, 112, 1094–1104.
- Bouchal, T.; Durník, I.; Kulhánek, P. Bending of Canonical and G/T Mismatched DNAs. J. Chem. Inf. Model. 2021, 61, 6000–6011.
- Sharma, M.; Predeus, A.V.; Mukherjee, S.; Feig, M. DNA Bending Propensity in the Presence of Base Mismatches: Implications for DNA Repair. J. Phys. Chem. B 2013, 117, 6194–6205.
- Maffeo, C.; Ngo, T.T.M.; Ha, T.; Aksimentiev, A. A Coarse-Grained Model of Unstructured Single-Stranded DNA Derived from Atomistic Simulation and Single-Molecule Experiment. J. Chem. Theory Comput. 2014, 10, 2891–2896.
- Rossetti, G.; Dans, P.D.; Gomez-Pinto, I.; Ivani, I.; Gonzalez, C.; Orozco, M. The structural impact of DNA mismatches. Nucleic Acids Res. 2015, 43, 4309–4321.
- Bouchal, T.; Durník, I.; Illík, V.; Réblová, K.; Kulhánek, P. Importance of base-pair opening for mismatch recognition. Nucleic Acids Res. 2020, 48, 11322–11334.
- Lavery, R.; Maddocks, J.H.; Pasi, M.; Zakrzewska, K. Analyzing ion distributions around DNA. Nucleic Acids Res. 2014, 42, 8138–8149.
- Tolokh, I.S.; Thomas, D.G.; Onufriev, A.V. Explicit ions/implicit water generalized Born model for nucleic acids. J. Chem. Phys. 2018, 148, 195101.
- Sun, L.Z.; Qian, J.L.; Cai, P.; Xu, X. Mutual effects between single-stranded DNA conformation and Na+-Mg2+ ion competition in mixed salt solutions. Phys. Chem. Chem. Phys. 2022, 24, 20867–20881.
- Xue, J.; Wang, P.; Li, X.; Tan, R.; Zong, W. Transformation characteristics of A-DNA in salt solution revealed through molecular dynamics simulations. Biophys. Chem. 2022, 288, 106845.
- Sarkar, S.; Singh, P.C. The combined action of cations and anions of ionic liquids modulates the formation and stability of G-quadruplex DNA. Phys. Chem. Chem. Phys. 2021, 23, 24497–24504.
- Pasi, M.; Maddocks, J.H.; Lavery, R. Analyzing ion distributions around DNA: Sequence-dependence of potassium ion distributions from microsecond molecular dynamics. Nucleic Acids Res. 2015, 43, 2412–2423.
- Fu, H.; Zhang, C.; Qiang, X.-W.; Yang, Y.-J.; Dai, L.; Tan, Z.-J.; Zhang, X.-H. Opposite Effects of High-Valent Cations on the Elasticities of DNA and RNA Duplexes Revealed by Magnetic Tweezers. Phys. Rev. Lett. 2020, 124, 058101.
- Cruz-Leon, S.; Vanderlinden, W.; Muller, P.; Forster, T.; Staudt, G.; Lin, Y.Y.; Lipfert, J.; Schwierz, N. Twisting DNA by salt. Nucleic Acids Res. 2022, 50, 5726–5738.
- Long, M.P.; Alland, S.; Martin, M.E.; Isborn, C.M. Molecular dynamics simulations of alkaline earth metal ions binding to DNA reveal ion size and hydration effects. Phys. Chem. Chem. Phys. 2020, 22, 5584–5596.
- Xi, K.; Wang, F.-H.; Xiong, G.; Zhang, Z.-L.; Tan, Z.-J. Competitive Binding of Mg2+ and Na+ Ions to Nucleic Acids: From Helices to Tertiary Structures. Biophys. J. 2018, 114, 1776–1790.
- Krüger, A.; Zimbres, F.M.; Kronenberger, T.; Wrenger, C. Molecular Modeling Applied to Nucleic Acid-Based Molecule Development. Biomolecules 2018, 8, 83.
- Oweida, T.J.; Kim, H.S.; Donald, J.M.; Singh, A.; Yingling, Y.G. Faculty Opinions recommendation of Assessment of AMBER Force Fields for Simulations of ssDNA. J. Chem. Theory Comput. 2021, 17, 1208–1217.
- Dans, P.D.; Ivani, I.; Hospital, A.; Portella, G.; González, C.; Orozco, M. How accurate are accurate force-fields for B-DNA? Nucleic Acids Res. 2017, 45, 4217–4230.
- Cruz-León, S.; Grotz, K.K.; Schwierz, N. Extended magnesium and calcium force field parameters for accurate ion–nucleic acid interactions in biomolecular simulations. J. Chem. Phys. 2021, 154, 171102.
- Castelli, M.; Doria, F.; Freccero, M.; Colombo, G.; Moroni, E. Studying the Dynamics of a Complex G-Quadruplex System: Insights into the Comparison of MD and NMR Data. J. Chem. Theory Comput. 2022, 18, 4515–4528.
- Havrila, M.; Stadlbauer, P.; Islam, B.; Otyepka, M.; Šponer, J. Effect of Monovalent Ion Parameters on Molecular Dynamics Simula-tions of G-Quadruplexes. J. Chem. Theory Comput. 2017, 13, 3911–3926.
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339.
- van Gunsteren, W.F.; Daura, X.; Hansen, N.; Mark, A.E.; Oostenbrink, C.; Riniker, S.; Smith, L.J. Validation of Molecular Simulation: An Overview of Issues. Angew. Chem. Int. Ed. 2018, 57, 884–902.
- Betz, R.M.; Dror, R.O. How Effectively Can Adaptive Sampling Methods Capture Spontaneous Ligand Binding? J. Chem. Theory Comput. 2019, 15, 2053–2063.
- Markthaler, D.; Fleck, M.; Stankiewicz, B.; Hansen, N. Exploring the Effect of Enhanced Sampling on Protein Stability Prediction. J. Chem. Theory Comput. 2022, 18, 2569–2583.
- Kasavajhala, K.; Lam, K.; Simmerling, C. Exploring Protocols to Build Reservoirs to Accelerate Temperature Replica Exchange MD Simulations. J. Chem. Theory Comput. 2020, 16, 7776–7799.



