Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yozo Mitsui | -- | 3889 | 2023-06-25 10:51:36 | | | |
| 2 | Camila Xu | Meta information modification | 3889 | 2023-06-25 11:10:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mitsui, Y.; Yamabe, F.; Hori, S.; Uetani, M.; Kobayashi, H.; Nagao, K.; Nakajima, K. Pathogenesis of Plaque in Peyronie’s Disease Cases. Encyclopedia. Available online: https://encyclopedia.pub/entry/46009 (accessed on 06 June 2026).
Mitsui Y, Yamabe F, Hori S, Uetani M, Kobayashi H, Nagao K, et al. Pathogenesis of Plaque in Peyronie’s Disease Cases. Encyclopedia. Available at: https://encyclopedia.pub/entry/46009. Accessed June 06, 2026.
Mitsui, Yozo, Fumito Yamabe, Shunsuke Hori, Masato Uetani, Hideyuki Kobayashi, Koichi Nagao, Koichi Nakajima. "Pathogenesis of Plaque in Peyronie’s Disease Cases" Encyclopedia, https://encyclopedia.pub/entry/46009 (accessed June 06, 2026).
Mitsui, Y., Yamabe, F., Hori, S., Uetani, M., Kobayashi, H., Nagao, K., & Nakajima, K. (2023, June 25). Pathogenesis of Plaque in Peyronie’s Disease Cases. In Encyclopedia. https://encyclopedia.pub/entry/46009
Mitsui, Yozo, et al. "Pathogenesis of Plaque in Peyronie’s Disease Cases." Encyclopedia. Web. 25 June, 2023.
Copy Citation
Peyronie’s disease (PD) is a benign condition caused by plaque formation on the tunica albuginea of the penis. It is associated with penile pain, curvature, and shortening, and contributes to erectile dysfunction, which worsens patient quality of life.
Peyronie’s disease
pathogenesis
risk factors
molecular mechanisms
1. Introduction
Peyronie’s disease (PD) is a connective tissue disorder characterized by the formation of fibrous lesions or plaques in the tunica albuginea (TA) of the penis. The early stage is an active phase, with affected individuals experiencing erectile pain and penile curvature onset. However, not all cases present with penile pain at onset, with the incidence ranging from 20% to 70% of reported cases [1]. At 12–18 months following PD onset, the disease transitions to a painless chronic phase, with a gradual worsening of penile curvature in 20–50% of the cases [1][2][3][4], during which changes in the penis rarely show spontaneous reversal. Therefore, a severely deformed penis may cause sexual intercourse disorders, such as difficulty with insertion, pain during intercourse, body posture restrictions, and erectile dysfunction (ED), which can adversely affect psychological well-being and quality of life (QoL). Notably, it has been reported that PD patients have mental distress and a distorted image of themselves, and depression is suspected in a high proportion [5][6][7]. Furthermore, a large-scale survey of more than 8000 PD patients conducted in Sweden showed that they had increased risk of self-harm, as well as anxiety and depression [8], while a recent study showed that younger patients tended to suffer more psychological and physical symptoms, as well as penile pain in the chronic phase of PD [9].
A large variety of medical treatments have been suggested and are utilized for PD. Nonsurgical treatments for affected patients include oral, topical, intralesional, extracorporeal shockwave, and traction therapy. In terms of oral treatment options, results have shown that a low dose of tadalafil may slow the progression of penile curvature in patients in the acute phase [10], while intralesional therapy with collagenase clostridium histolyticum is reported to be capable of improving penile curvature [11]. Additionally, extracorporeal shockwave therapy (ESWT) can cause plaque damage, resulting in plaque resorption in some affected patients [12][13]. In cases of chronic PD, assistive devices (penile traction, vacuum devices, penile prostheses) may be effective for improving penile curvature [14][15]. When these treatments are ineffective, surgical treatment such as a grafting procedure is often attempted to correct penile curvature [16].
Although the exact etiology of PD remains unknown, evidence accumulated over the past two decades shows several molecular alterations and aberrant signaling pathways involved in the pathogenesis. In addition, epidemiological studies have indicated various risk factors possibly associated with its development, such as smoking, hypertension (HT), diabetes mellitus (DM), and older age [17][18]. Possible connections to certain genetic predispositions have also been proposed as being involved in PD development, including single nucleotide polymorphisms (SNPs) although the exact susceptibility factors have yet to be established [19][20][21][22]. The prevalence of PD has been reported to range from 0.6% to 20%, with wide variations among regions and ethnic groups although it is particularly low in Asians [22][23][24][25][26][27].
2. Pathogenesis of Plaque in PD Cases
The hypothesis posed by Devine et al. that PD is initiated by repetitive micro-trauma to the penis during intercourse has become widely accepted [28]. In one study, rats that received repeated local injections of chlorhexidine ethanol to produce repetitive microtrauma to the TA developed fibrous plaque in the penis [29]. For affected patients, the primary site of injury is thought to be the TA, with disruption of the inner and outer layers creating a milieu for inflammation, elastic fiber disruption, and extravascular blood accumulation [21]. This serves as a scaffold for fibrin and fibronectin accumulation, which is followed by infiltration of inflammation-associated cells that release various types of cytokines and subsequently show cell-type conversion, including tissue-resident fibroblasts and smooth muscle cells, into myofibroblasts characterized by α-smooth muscle actin (α-SMA) expression and collagen secretion. Transforming growth factor-β1 (TGF-β1) is a key factor for myofibroblast activation and a major player in fibrosis in all organs [19][20][21][30]. Myofibroblasts are a crucial element for the fibrotic process, as they cause excessive accumulation of collagen fibers, fibronectin, and other components of the extracellular matrix (ECM). An imbalance between matrix metalloproteinase (MMP), which removes collagen fibers, and its tissue inhibitor of metalloproteinase (TIMP) in myofibroblasts, along with the evasion of apoptosis, are thought to be key factors in the development of fibrotic diseases [21][31][32]. Interestingly, recent studies have revealed the involvement of persistent immunological features including mainly macrophages with fibrosis including PD [20][32][33][34][35][36][37]. Based on the latest reported findings, details regarding the mechanisms of fibrotic tissue remodeling and putative functions of each cell type during different stages of PD are presented below and in Figure 1.
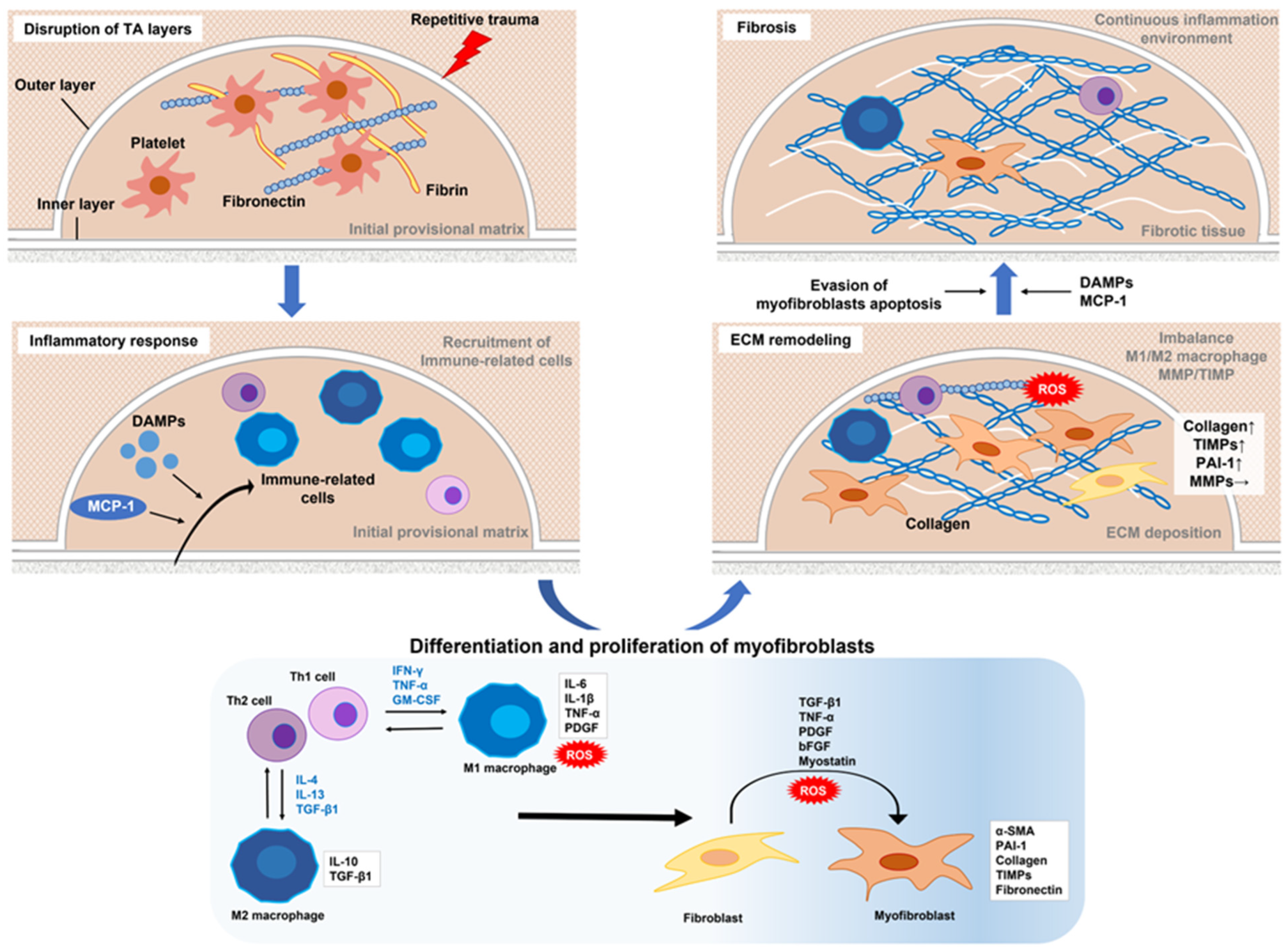
Figure 1. Proposed mechanism of molecular regulation of Peyronie’s disease. Repeated microtrauma disrupts the inner and outer layers of the TA. Platelets are activated and then release granules that form an early provisional matrix composed of fibrin and fibronectin. In addition, DAMPs and MCP-1 released from various cells upon tissue damage or mechanical stress promote the recruitment of inflammation-related cells to the early provisional matrix. Th1 cells produce IFN-γ, TNF-α, and GM-CSF to activate M1 macrophages, and Th2 cells produce IL-4, IL-13, and TGF-β1 to activate M2 macrophages. Activated M1 macrophages exhibit increased antigen-presenting activity and the high production of pro-inflammatory cytokines, such as IL-6, IL-1b, and TNF-α, as well as ROS. M2 macrophages are characterized by increased expressions of the anti-inflammatory cytokine IL-10 and profibrotic factor TGF-β1. An M1–M2 imbalance, expressed by excessive M2 activity, induces fibroblast activation (conversion to myofibroblasts) and promotes fibrosis. Conversion to myofibroblasts involves TGF-β1, TNF-α, IL-6, PDGF, bFGF, myostatin, and ROS. Activated myofibroblasts produce collagen and fibronectin, components of the ECM. On the other hand, the expression of MMPs that cause ECM degradation is largely unchanged by the overproduction of PAI-1 and TIMP, resulting in excessive accumulation of ECM. Increased ROS due to excessive ECM deposition, which promotes release of DAMPs due to tissue damage, and continued recruitment and stimulation of immune-related cells by MCP-1 released from myofibroblasts can lead to sustained activation of myofibroblasts. Furthermore, evasion of myofibroblast apoptosis is also critically involved in the maintenance of a chronic extracellular fiber environment. As a result, ECM continues to accumulate and is thought to promote fibrosis. TA–tunica albuginea; DAMPs–damage-associated molecular patterns; MCP-1–monocyte chemoattractant protein 1; IFN-γ–interferon gamma; TNF-α, tumor necrosis factor-alpha; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL–interleukin; TGF-β1–transforming growth factor-β1; ROS–reactive oxygen species; PDGF–platelet-derived growth factor; bFGF–basic fibroblast growth factor; ECM–extracellular matrix; MMPs–matrix metalloproteinases; PAI-1–plasminogen activator inhibitor type 1; TIMP–tissue inhibitor of metalloproteinase.
2.1. Role of the Immune System in Inflammatory Response
In the tissue fibrosis process, innate and adaptive immune systems are both intricately involved in inflammatory events. Macrophages and monocytes are key immune-related cells related to the innate immune system, while T cells and B cells are involved in the adaptive immune system.
At the site of injury, platelets become activated, then release granules and stop blood loss first by forming an initial provisional matrix composed of fibrin and fibronectin, which has also been identified in PD [38]. Fibrin and fibronectin also function to alter leukocyte recruitment to the wound site and promote fibrosis [39]. The chemokine monocyte chemoattractant protein 1 (MCP-1), another factor that mediates recruitment and activation of macrophages and monocytes, is produced and secreted from monocytes/macrophages, fibroblasts, and vascular endothelial cells by stimulation of lipopolysaccharide and inflammatory cytokines upon tissue injury [40][41]. A gene expression profile study using PD-derived lesions demonstrated significantly elevated expression of MCP-1 in PD plaque over a normal TA condition [42], while Lin et al. confirmed higher levels of MCP-1 mRNA in PD than in non-PD cells [43]. Other factors involved in recruitment and stimulation of immune-related cells include pathogen-associated molecular patterns and damage-associated molecular patterns (DAMPs), which stimulate through various receptors on immune cells such as toll-like receptors (TLR) (mostly-2 and -4) [20][37][44]. DAMPs are released following stress or injury to various cells, including dead cells, proliferating neutrophils, macrophages, lymphocytes, natural killer cells, mesenchymal stem cells (MSCs), and resident cells [45][46][47]. In 2000, Mills et al. proposed a new classification for macrophages as either M1—pro-inflammatory— or M2—anti-inflammatory/profibrotic [48]. Thereafter, M2 macrophages were further classified into M2a, M2b, M2c, and M2d subtypes based on the secretion of distinct cytokines, the presence of certain cell surface proteins, gene expression profiles, and other biological activities, while they commonly express interleukin (IL)-10 [49][50].
There are different subtypes of T cells, each with unique regulatory effects on inflammation and fibrosis, including through cytokine release. Among these, helper T cells are a subpopulation of lymphocytes that express the CD4 antigen on the cell surface and are now mainly divided into Th1, Th2, and Th17 types [51]. Th1 cells differentiate generally in the presence of IL-12 and mainly produce interferon gamma (IFN-γ) after differentiation, whereas Th2 cells differentiate in the presence of IL-4 and continue to produce mainly IL-4 after differentiation, with each having an influence on macrophage activation [52]. In addition to granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor-alpha (TNF-α), the Th1 cell cytokine IFN-γ, activates polarization of M1 macrophages via various pathways, including the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and nuclear factor (NF)-κB pathways [36][37][53]. The Th2 cytokine IL-4, similar to IL-13 and TGF-β1, activates M2 macrophages through STAT6 or interferon regulatory factor 4-mediated signaling [54][55][56], with this macrophage plasticity being known as M1/M2 polarization. M1 macrophages activated in this manner exhibit high levels of antigen-presenting activity and production of pro-inflammatory cytokines such as IL-6 and TNF-α, as well as the production of nitric oxide (NO) and reactive oxygen species (ROS) [36][37][55]. In contrast, M2 macrophages produce fewer of the inflammatory cytokines IL-6 and TNF-α. In particular, the M2c type is characterized by high expression levels of the anti-inflammatory cytokine IL-10 and profibrotic factor TGF-β1, thus contributing to tissue remodeling and fibrosis promotion [36][37][55][56][57].
B cells contribute to several different fibrotic diseases via a number of mechanisms, including direct cell–cell contact and the production of profibrotic and pro-inflammatory cytokines (e.g., IL-6 and TGF-β1) [58][59][60]. Furthermore, decreased IL-10 production by B cells is possibly a factor associated with fibrosis according to previous examinations of systemic sclerosis patients [60][61][62]. These studies also showed that increased IL-6 and reduced IL-10 levels in B cells are induced through the activation of the JAK/STAT and TLRs pathways.
M1/M2 macrophage phenotypes are interchangeable depending on the microenvironment, while an M1–M2 imbalance expressed by excessive M2 activity is associated with different types of fibrosis through excessive fibroblast activation [36][63][64]. Using a cisplatin-induced rat renal fibrosis model, Nakagawa et al. showed that the number of M1 macrophages begin to increase with the upregulation of M1-related cytokines (IFN-γ, TNF-α, IL-6) in the middle stage and the decrease in the late stage [65]. In contrast, M2 macrophages exhibit progressively increased numbers in the middle and late stages, accompanied by increased profibrotic factor TGF-β1 expression. This study also found increased numbers of CD4+ and CD8+ T cells in the late stage of the fibrotic process. Thus, M1 macrophages are involved in the relatively early stages of wound response and are thought to be replaced at later stages by specific repair populations of M2 macrophages via T–cell responses. Regarding PD, serum IL-6 levels were found to be significantly elevated in patients with PD in the acute phase as compared to healthy controls [66]. In contrast, a study that compared 91 PD patients with healthy controls showed that patients in the chronic stage had elevated serum levels of TGF-β1 and IFN-γ, and decreased levels of TNF-α, while IL-6 was undetectable [67]. In addition, a recent study by Cui et al. that used integrated bioinformatic analysis provided interesting findings regarding the association of PD with immune-related cells [68]. Research based on immune cell infiltration analysis demonstrated that various immune-related cells, such as T cells, B cells, macrophages, neutrophils, and plasma cells, are widely involved in fibrosis in PD patients. Additionally a number of genes were also identified, including inflammatory cytokines such as IL-6, as being important immune-related factors in PD and their correlations were shown. Notably, functional enrichment analysis showed that these genes have potential regulatory roles in several key biological processes, including the JAK/STAT pathway. Thus, complicated mechanisms that arise through M1–M2 macrophage interactions and T–cell responses may contribute to PD development.
2.2. Differentiation and Proliferation of Myofibroblasts in the Profibrotic Environment
Myofibroblasts, which function in tissue contraction and ECM secretion, have an important role during impaired healing. The profibrotic milieu, formed by the infiltration and activation of leukocytic tissue, as well as Th2 cell–M2 macrophage polarization, induces mesenchymal transition (conversion to myofibroblasts) in various cell types, such as tissue-resident fibroblasts, fibrocytes of different origin, smooth muscle and endothelial cells, and mesenchymal stem cells [21]. Various factors are involved in conversion to myofibroblasts, among which TGF-β1 plays a central role. Others include pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β), growth factors [e.g., platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)], myostatin, and ROS.
PDGF is primarily secreted by endothelial cells and macrophages, and is also released from platelets upon degranulation, with its production as well as expression of related receptors being maintained by TGF-β1 signaling [20]. Hamid et al. showed that pro–inflammatory macrophages promote the myofibroblast transformation of cardiac MSCs through the activation of PDGF and its receptor PDGFR-β [69], while another study using a rat PD model revealed significantly elevated expression levels of PDGF-α and PDGF-β as well as their receptors PDGFR-α and PDGFR-β [70]. Interestingly, a recent study that used single-cell RNA sequencing (scRNA-Seq) with samples from patients with Dupuytren’s disease (DD), widely known to be associated with PD, showed that the endothelium functions to promote an immune regulatory fibroblast phenotype through PDGF signaling during fibrosis development [71]. bFGF, one of the factors that is expressed in the early stage of tissue injury and that plays an important role in wound healing [72][73], not only functions as a fibroblast mitogen, but also promotes myofibroblast proliferation and migration after TGF-β1-induced mesenchymal transition [4]. It was also shown that production of bFGF by PD plaque-derived myofibroblasts is significantly greater than that of by normal TA myofibroblasts [74]. Furthermore, myostatin, a member of the TGF-β family, may promote fibrous tissue formation in muscle tissue by transforming fibroblasts into myofibroblasts to replace damaged muscle fibers [75]. A study of the effect of myostatin deficiency on alterations in left ventricular function after myocardial infarction showed that α-SMA activity was less intense in myostatin knockout than in wild-type mice, suggesting the possibility of fewer myofibroblasts [76]. Moreover, myostatin was found to be overexpressed in PD plaque and to stimulate myofibroblast generation and collagen expression in rat TA cells [77].
Accumulation of ROS is induced by hypoxia and produces oxidative stress, which has detrimental effects on various cellular components. ROS-induced myofibroblast transformation involves a pathway through ROS-mediated HIF-1α stabilization [78][79]. For example, HIF-1α was shown to induce abnormal fibroblast activity via the activation of the extracellular signal-related kinase signaling pathway in cutaneous wound healing [80]. Another study found that rats with PD-like lesions expressed high levels of HIF-1α and inducible NO synthase (iNOS) via the activation of NF-κB [70]. More recently, results of bioinformatic approaches identified a number of potential hypoxia-associated genes associated with PD [81].
2.3. Extracellular Matrix Deposition
Following an initial inflammatory event, fibrosis progression is facilitated by the accumulation of ECM components secreted by myofibroblasts, leading to fibrotic tissue remodeling. The predominant components include collagen, especially types I, III, and IV, in penile plaque, proteoglycans, fibronectin, and fibromodulin [19][82][83], with excessive accumulation caused by disruption of the complex interactions of various factors.
MMPs, neutral proteases, play a central role in ECM degradation and when activated lead to ECM degradation during normal wound healing. Their activation is regulated by various factors, including TGF-β1, PDGFs, IL-1, TNF-α, and bFGF. For example, TGF-β1 activates collagenase MMPs (MMP-1, -8, and -13, which degrade types I, II, and III collagen) and gelatinase MMPs (MMP-2 and -9, which degrade types IV, V, and VII collagen, as well as gelatin and elastin) [84]. However, a previous study found that administration of TGF-β1 to a fibroblast cell line established from PD plaque did not alter MMP-1, -8, or -13 expressions and only mildly increased that of MMP-9, although IL-1β induced production of MMP-1, -3, -10, and -13 [85]. Another study compared samples from PD plaque and normal TA using immunostaining and also found no differences in MMP-2 or -9 expression between them [86]. Instead, this study confirmed that administration of TGF-β1 induced excessive accumulation of TIMP1-4, which suppresses the activity of MMPs, in fibroblast cell lines established from PD plaque [85]. TIMPs are mainly produced in macrophages, fibroblasts, and myofibroblasts, and their expression is also positively regulated by PDGF and bFGF. Plasminogen activator inhibitor type 1 (PAI-1), synthesized in various cell types including endothelial cells, fibroblasts, and macrophages, is also known to inhibit plasmin-mediated MMP activation and promote fibrosis [87]. Davila et al. found that both mRNA and protein levels of PAI-1 were significantly increased in human PD plaque samples and respective fibroblast cultures as compared to normal TA [88]. Therefore, excessive ECM accumulation during PD plaque development may be driven by altered MMP levels caused by the inhibitory effects of TIMP and PAI-1 on MMP activation.
2.4. Progression of Fibrosis in Environment with Continuous Inflammation
In normal wound healing, myofibroblasts contract via the increased expression of matrix protein receptor integrins to regulate excessively deposited ECM and then continue to support mechanical loads until collagen cross-linking for formation of striated scar tissue [39]. After the wound is repaired, myofibroblasts undergo apoptosis, and the repair response is terminated [82]. However, in fibrotic diseases including PD, tissue remodeling and fibroblast activation persist as chronic, uncontrolled processes, with structural changes in fibrotic tissue thought to play an important role in the formation of this chronic extracellular profibrotic milieu [89]. Progressive deposition of ECM proteins creates a hypoxic environment by increasing stiffness around the affected TA and inhibiting oxygen diffusion. Lack of oxygen, the terminal electron acceptor of the electron transport chain, increases ROS generation, further promoting cellular and tissue damage and myofibroblast activation [81][90]. ROS-induced damage then induces release of DAMPs from damaged cells, resulting in further continued recruitment and stimulation of macrophages and lymphocytes. Continuous recruitment and stimulation of immune-related cells is also maintained by fibronectin and MCP-1 released from TGF-β1-activated myofibroblasts [91]. Thus, structural changes in fibrous tissue due to excessive ECM deposition may lead to sustained myofibroblast activation, contributing to continuation of the profibrotic milieu.
The effect of ESWT on PD in the early phase is thought to be related to direct destruction of the penile plaque by shockwaves, as well as plaque lysis caused by the activation of macrophages induced by an inflammatory response to heat generated by the treatment [92]. However, as noted above, macrophage activation is a major factor that promotes fibrosis, and the clinical significance of ESWT remains questionable. A recently published systematic review and meta-analysis indicates that ESWT may reduce plaque size, although it fails to improve penile curvature or pain in PD patients [13].
Evasion of myofibroblast apoptosis is also critically involved in maintaining a chronic extracellular fibrous milieu. Various types of programmed cell death, including apoptosis, are known to be closely associated with organ fibrosis [93]. Apoptotic pathways include intrinsic pathways activated by cellular stress and DNA damage–as among other causes–and extrinsic pathways activated via the detection of extracellular death signals from other cells. The intrinsic apoptotic pathway is tightly regulated by a balance of the activities of Bcl-2 family proteins, consisting of pro–apoptotic (Bax, Bak, Bad, Bid, Puma, Bim, Noxa) and anti–apoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1) proteins. Furthermore, both intrinsic and extrinsic apoptotic pathways ultimately depend on the protease activity of the initiator (e.g., caspase-2, -8, -9, -10, -12) and executioner (e.g., caspase-3, -6, -7). Interestingly, Zorba et al. found high levels of Bcl-2 expression and decreased levels of caspase-3 and -8 expression in PD plaque, indicating apoptosis suppression [94]. In contrast, Loreto et al. showed that PD tissues had a lower expression of Bcl-2 but an overexpression of Bax and caspase-3 and -9 as compared to normal TA [95]. They also reported that a tumor necrosis factor-related apoptosis-inducing ligand and its receptor DR5 were overexpressed in PD tissues, suggesting that apoptosis may be induced by intrinsic and extrinsic pathways [96]. These different results may be partially explained by a new concept of apoptosis evasion in myofibroblasts termed “mitochondrial priming” [97], which refers to the proximity of mitochondria to the apoptotic threshold and which is determined by the relative expression of pro–apoptotic and pro–survival members of the Bcl-2 family of proteins. Myofibroblasts are “primed” and thus ready to die at the time of conversion from fibroblasts and become dependent on pro–survival proteins to sequester pro–apoptotic proteins and ensure survival. Therefore, various types of apoptosis-associated proteins are expressed at different ratios in the mitochondria of myofibroblasts that continue to survive while avoiding apoptosis, which might be a factor contributing to differences in these results. Additionally, epigenetic modifications may lead to sustained myofibroblast activation and consequent fibrosis, the details of which will be discussed later.
PD plaques formed in this way evolve toward calcification in 20–25% of affected cases [98]. PDGF functions as an osteoblast recruiter and contributes to calcification and ossification of PD plaques [70]. Following the initial inflammatory phase, the disease moves to the second phase (chronic), during which time the disease stabilizes. The absence of pain and inflammation has long been thought to be a hallmark of the chronic phase. However, a recent study by Milenkovic et al. that used immunohistochemistry and scRNA-Seq indicated the possibility of a sustained inflammatory reaction even in the chronic PD stage [99]. Such findings showing inflammation in PD may pave the way for the development of new treatments for patients in the chronic phase. A brief summary of the key molecules and their roles in the pathophysiology of PD is presented in Table 1.
Table 1. Summary of molecules associated with development of Peyronie’s disease and their roles.
| Associated Molecules | Production Sources | Upregulators/Activators | Physiological Activities |
|---|---|---|---|
| MCP-1 | Monocytes, macrophages, fibroblasts, | Lipopolysaccharide, | Recruitment and activation of macrophages and monocytes |
| vascular endothelial cells | inflammatory cytokines | ||
| DAMPs | Dead cells, proliferating neutrophils, | Stress, tissue injury, ROS | Recruitment and activation of immune-related cells through toll-like receptors |
| macrophages, lymphocytes, | |||
| natural killer cells, resident cells, | |||
| mesenchymal stem cells | |||
| IFN-γ | Th1 cells | IL-12 | Activation of M1 macrophage polarization |
| TNF-α | Th1 cells, macrophages (M1) | IL-12 | Activation of M1 macrophage polarization, |
| promotion of differentiation of myofibroblasts | |||
| GM-CSF | Th1 cells | IL-12 | Activation of M1 macrophage polarization |
| IL-6 | Macrophages (M1) | IFN-γ, TNF-α, GM-CSF | Promotion of differentiation of myofibroblasts |
| TGF-β1 | Platelets, fibroblasts, Th2 cells, | Th2 cells, PAI-1, integrin, | Promotion of differentiation of myofibroblasts |
| macrophages (M2) | thrombospondin 1, plasmin, | ||
| MMP-2, -9, ROS | |||
| PDGF | Endothelial cells, macrophages (M1), | TGF-β1 | Promotion of differentiation of myofibroblasts |
| platelets | |||
| bFGF | Various cell types | Tissue injury | Promotion of differentiation of myofibroblasts |
| and fibroblast mitogen | |||
| Myostatin | Skeletal muscle | Promotion of differentiation of myofibroblasts | |
| ROS | Macrophages (M1), various cells | Hypoxia, inflammation | Promotion of differentiation of myofibroblasts through |
| ROS-mediated HIF-1α stabilization | |||
| MMPs | Myofobroblasts | TGF-β1, PDGF, TNF-α, bFGF | Extracellular matrix degradation |
| TIMPs | Macrophages, fibroblasts, myofibroblasts | TGF-β1 | Suppression of MMP activity |
| PAI-1 | Various cell types | TGF-β1, collagen | Inhibition of plasmin-mediated MMP activation |
Abbreviations: MCP-1—monocyte chemoattractant protein 1; DAMPs—damage-associated molecular patterns; IFN-γ—interferon gamma; TNF-α—tumor necrosis factor-alpha; GM-CSF—granulocyte-macrophage colony-stimulating factor; IL-6, 12—interleukin-6, 12; TGF-β1—transforming factor-beta 1; PDGF—platelet-derived growth factor; bFGF—basic fibroblast growth factor; ROS—reactive oxygen species; MMP—matrix metalloproteinase; TIMPs—tissue inhibitor of metalloproteinases; HIF-1α—hypoxia inducible factor-1 alpha; PAI-1—plasminogen activator inhibitor-1.
References
- Di Maida, F.; Cito, G.; Lambertini, L.; Valastro, F.; Morelli, G.; Mari, A.; Carini, M.; Minervini, A.; Cocci, A. The natural history of Peyronie’s disease. World J. Mens Health 2021, 39, 399–405.
- Kadioglu, A.; Tefekli, A.; Erol, B.; Oktar, T.; Tunc, M.; Tellaloglu, S. A retrospective review of 307 men with Peyronie’s disease. J. Urol. 2002, 168, 1075–1079.
- Mulhall, J.P.; Schiff, J.; Guhring, P. An analysis of the natural history of Peyronie’s disease. J. Urol. 2006, 175, 2115–2118.
- Berookhim, B.M.; Choi, J.; Alex, B.; Mulhall, J.P. Deformity stabilization and improvement in men with untreated Peyronie’s disease. BJU. Int. 2014, 113, 133–136.
- Nelson, C.J.; Mulhall, J.P. Psychological impact of Peyronie’s disease: A review. J. Sex. Med. 2013, 10, 653–660.
- Terrier, J.E.; Nelson, C.J. Psychological aspects of Peyronie’s disease. Transl. Androl. Urol. 2016, 5, 290–295.
- Nelson, C.J.; Diblasio, C.; Kendirci, M.; Hellstrom, W.; Guhring, P.; Mulhall, J.P. The chronology of depression and distress in men with Peyronie’s disease. J. Sex. Med. 2008, 5, 1985–1990.
- Kuja-Halkola, R.; Henningsohn, L.; D’Onofrio, B.M.; Mills, J.; Adolfsson, A.; Larsson, H.; Cederlöf, M. Mental disorders in Peyronie’s disease: A Swedish cohort study of 3.5 million men. J. Urol. 2021, 205, 864–870.
- Cilio, S.; Fallara, G.; Capogrosso, P.; Candela, L.; Belladelli, F.; Pozzi, E.; Corsini, C.; Raffo, M.; Schifano, N.; Boeri, L.; et al. The symptomatic burden of Peyronie’s disease at presentation according to patient age: A critical analysis of the Peyronie’s disease questionnaire (PDQ) domains. Andrology 2023, 11, 501–507.
- Spirito, L.; Manfredi, C.; La Rocca, R.; Napolitano, L.; Di Girolamo, A.; Capece, M.; Trama, F.; Sciorio, C.; Sokolakis, I.; Creta, M.; et al. Daily low dose tadalafil may reduce the penile curvature progression rate in patients with acute Peyronie’s disease: A retrospective comparative analysis. Int. J. Impot. Res. 2022.
- Capece, M.; Arcaniolo, D.; Manfredi, C.; Palmieri, A.; De Sio, M.; Verze, P.; Fusco, F.; Longo, N.; Mirone, V. Second cycle of intralesional Collagenase Clostridium histolyticum for Peyronie’s disease using the modified shortened protocol: Results from a retrospective analysis. Andrologia 2020, 52, e13527.
- Manfredi, C.; Arcaniolo, D.; Amicuzi, U.; Spirito, L.; Napolitano, L.; Crocerossa, F.; Paoletta, M.; Gisone, S.; Cirillo, P.; Crocetto, F.; et al. Impact of extracorporeal shockwave therapy for erectile dysfunction and Peyronie’s disease on reproductive and hormonal testicular function. Andrology 2022, 10, 1368–1375.
- Bakr, A.M.; El-Sakka, A.I. Extracorporeal shockwave therapy in Peyronie’s disease: Systematic review and meta-analysis. J. Sex. Med. 2021, 18, 1705–1714.
- Krishnappa, P.; Manfredi, C.; Sinha, M.; Arcaniolo, D.; Matippa, P.; Moncada, I. Penile modeling in Peyronie’s disease: A systematic review of the literature. Sex. Med. Rev. 2022, 10, 434–450.
- Verze, P.; Sokolakis, I.; Manfredi, C.; Collà Ruvolo, C.; Hatzichristodoulou, G.; Romero-Otero, J. Penile prosthesis implant in the management of Peyronies’ disease. Minerva. Urol. Nephrol. 2021, 73, 196–214.
- Fernández-Pascual, E.; Manfredi, C.; Torremadé, J.; Ibarra, F.P.; Geli, J.S.; Romero-Otero, J.; García-Baquero, R.; Poblador, A.F.; Barbará, M.R.; Campos-Juanatey, F.; et al. Multicenter prospective study of grafting with collagen fleece TachoSil in patients with Peyronie’s disease. J. Sex. Med. 2020, 17, 2279–2286.
- Gelbard, M.K.; Rosenbloom, J. Fibroproliferative disorders and diabetes: Understanding the pathophysiologic relationship between Peyronie’s disease, Dupuytren disease and diabetes. Endocrinol. Diabetes. Metab. 2020, 4, e00195.
- Segundo, A.; Glina, S. Prevalence, risk factors, and erectile dysfunction associated with Peyronie’s disease among men seeking urological care. Sex. Med. 2020, 8, 230–236.
- Herati, A.S.; Pastuszak, A.W. The genetic basis of Peyronie disease: A review. Sex. Med. Rev. 2016, 4, 85–94.
- Milenkovic, U.; Ilg, M.M.; Cellek, S.; Albersen, M. Pathophysiology and future therapeutic perspectives for resolving fibrosis in Peyronie’s disease. Sex. Med. Rev. 2019, 7, 679–689.
- Krakhotkin, D.V.; Chernylovskyi, V.A.; Mottrie, A.; Greco, F.; Bugaev, R.A. New insights into the pathogenesis of Peyronie’s disease: A narrative review. Chronic. Dis. Transl. Med. 2020, 6, 165–181.
- Rhoden, E.L.; Teloken, C.; Ting, H.Y.; Lucas, M.L.; Teodosio da Ros, C.; Ary Vargas Souto, C. Prevalence of Peyronie’s disease in men over 50 years old in southern Brazil. Int. J. Impot. Res. 2001, 13, 291–293.
- Tal, R.; Heck, M.; Teloken, P.; Siegrist, T.; Nelson, C.J.; Mulhall, J.P. Peyronie’s disease following radical prostatectomy: Incidence and predictors. J. Sex. Med. 2010, 7, 1254–1261.
- Shiraishi, K.; Shimabukuro, T.; Matsuyama, H. The prevalence of Peyronie’s disease in Japan: A study in men undergoing maintenance hemodialysis and routine health checks. J. Sex. Med. 2012, 9, 2716–2723.
- Chung, E.; Ralph, D.; Kagioglu, A.; Garaffa, G.; Shamsodini, A.; Bivalacqua, T.; Glina, S.; Hakim, L.; Sadeghi-Nejad, H.; Broderick, G. Evidence-based management guidelines on Peyronie’s disease. J. Sex. Med. 2016, 13, 905–923.
- Capoccia, E.; Levine, L.A. Contemporary review of Peyronie’s disease treatment. Curr. Urol. Rep. 2018, 19, 51.
- Cocci, A.; Russo, G.I.; Briganti, A.; Salonia, A.; Cacciamani, G.; Capece, M.; Falcone, M.; Timpano, M.; Cito, G.; Verze, P.; et al. Predictors of treatment success after collagenase Clostridium histolyticum injection for Peyronie’s disease: Development of a nomogram from a multicentre single-arm, non-placebo controlled clinical study. BJU Int. 2018, 122, 680–687.
- Devine, C.J., Jr.; Somers, K.D.; Jordan, S.G.; Schlossberg, S.M. Proposal: Trauma as the cause of the Peyronie’s lesion. J. Urol. 1997, 157, 285–290.
- Jiang, H.; Gao, Q.; Che, X.; Zhu, L.; Zhang, Z.; Chen, Y.; Dai, Y. Repeated micro-trauma of the penile tunica albuginea: A new animal model of Peyronie’s disease. Urol. Int. 2018, 100, 228–239.
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053.
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37.
- Milenkovic, U.; Ilg, M.; Cellek, S.; Albersen, M.E. What role do pharmaceuticals play in the treatment of Peyronie’s disease and is there a need for new emerging drugs? Expert Opin. Emerg. Drugs 2019, 24, 1–4.
- O’Dwyer, D.N.; Ashley, S.L.; Moore, B.B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L590–L601.
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89.
- Wu, L.; Huang, K.; Li, Q.; Wu, H.; Gao, Y.; Xu, X.; Liu, X.; Han, L. Crosstalk between myofibroblasts and macrophages: A regulative factor of valvular fibrosis in calcific aortic valve disease. Cell Biol. Int. 2023, 47, 754–767.
- Lis-López, L.; Bauset, C.; Seco-Cervera, M.; Cosín-Roger, J. Is the macrophage phenotype determinant for fibrosis development? Biomedicines 2021, 9, 1747.
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730.
- Van de Water, L. Mechanisms by which fibrin and fibronectin appear in healing wounds: Implications for Peyronie’s disease. J. Urol. 1997, 157, 306–310.
- Moretti, L.; Stalfort, J.; Barker, T.H.; Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022, 298, 101530.
- Jiang, Y.; Beller, D.I.; Frendl, G.; Graves, D.T. Monocyte chemoattractant protein 1 regulates adhesion molecule expression and cytokine production in human monocytes. J. Immunol. 1992, 148, 2423–2438.
- Young, L.R.; Gulleman, P.M.; Short, C.W.; Tanjore, H.; Sherrill, T.; Qi, A.; McBride, A.P.; Zaynagetdinov, R.; Benjamin, J.T.; Lawson, W.E.; et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight 2016, 1, e88947.
- Magee, T.R.; Qian, A.; Rajfer, J.; Sander, F.C.; Levine, L.A.; Gonzalez-Cadavid, N.F. Gene expression profiles in the Peyronie’s disease plaque. Urology 2002, 59, 451–457.
- Lin, C.S.; Lin, G.; Wang, Z.; Maddah, S.A.; Lue, T.F. Upregulation of monocyte chemoattractant protein 1 and effects of transforming growth factor-beta 1 in Peyronie’s disease. Biochem. Biophys. Res. Commun. 2002, 295, 1014–1019.
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337.
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040.
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276.
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 373, 96.
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. AMJ. Immunol. 2000, 164, 6166–6173.
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686.
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators. Inflamm. 2015, 2015, 816460.
- Zielinski, C.E. T helper cell subsets: Diversification of the field. Eur. J. Immunol. 2023, 15, e22.
- Teixeira, F.F.C.; Cardoso, F.G.R.; Ferreira, N.S.; Corazza, B.J.M.; Valera, M.M.C.; Nascimento, G.G.; Martinho, F.C. Effects of calcium hydroxide intracanal medications on T helper (Th1, Th2, Th9, Th17, and Tfh) and regulatory T (Treg) cell cytokines in apical periodontitis: A CONSORT RCT. J. Endod. 2022, 48, 975–984.
- Huang, X.; Li, Y.; Fu, M.; Xin, H.B. Polarizing macrophages in vitro. Methods Mol. Biol. 2018, 1784, 119–126.
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 2019, 10, 792.
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896.
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801.
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358.
- Todd, N.W.; Scheraga, R.G.; Galvin, J.R.; Iacono, A.T.; Britt, E.J.; Luzina, I.G.; Burke, A.P.; Atamas, S.P. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J. Inflamm. Res. 2013, 6, 63–70.
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res. Ther. 2018, 20, 75.
- Beesley, C.F.; Goldman, N.R.; Taher, T.E.; Denton, C.P.; Abraham, D.J.; Mageed, R.A.; Ong, V.H. Dysregulated B cell function and disease pathogenesis in systemic sclerosis. Front. Immunol. 2023, 13, 999008.
- Taher, T.E.; Ong, V.H.; Bystrom, J.; Hillion, S.; Simon, Q.; Denton, C.P.; Pers, J.O.; Abraham, D.J.; Mageed, R.A. Association of defective regulation of autoreactive interleukin-6-producing transitional B lymphocytes with disease in patients with systemic sclerosis. Arthritis. Rheumatol. 2018, 70, 450–461.
- Forestier, A.; Guerrier, T.; Jouvray, M.; Giovannelli, J.; Lefèvre, G.; Sobanski, V.; Hauspie, C.; Hachulla, E.; Hatron, P.Y.; Zéphir, H.; et al. Altered B lymphocyte homeostasis and functions in systemic sclerosis. Autoimmun. Rev. 2018, 17, 244–255.
- Ucero, A.C.; Bakiri, L.; Roediger, B.; Suzuki, M.; Jimenez, M.; Mandal, P.; Braghetta, P.; Bonaldo, P.; Paz-Ares, L.; Fustero-Torre, C.; et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J. Clin. Invest. 2019, 129, 3293–3309.
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295.
- Nakagawa, M.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunophenotypical characterization of M1/M2 macrophages and lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells 2021, 10, 257.
- Atar, A.; Kural, A.; Yenice, G.; Comez, I.; Tugcu, V.K. Role of interleukin-6 and pentraxin 3 as an early marker in Peyronie’s disease. Kaohsiung J. Med. Sci. 2017, 33, 195–200.
- Zimmermann, R.P.; Feil, G.; Bock, C.; Hoeltl, L.; Stenzl, A. Significant alterations of serum cytokine levels in patient with Peyronie’s disease. Int. Braz. J. Urol. 2008, 34, 457–466; discussion 466.
- Cui, Y.; Chen, L.; Wang, X.; Yu, L.; Wu, J. Identifying hub genes, key pathways and key immune-related genes in Peyronie’s disease by integrated bioinformatic analysis. Front. Pharmacol. 2022, 13, 1019358.
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Rokosh, G.; Jinno, M.; Zhou, G.; Wang, Q.; Prabhu, S.D. Cardiac mesenchymal stem cells promote fibrosis and remodeling in heart failure: Role of PDGF signaling. JACC Basic. Transl. Sci. 2022, 7, 465–483.
- Lucattelli, M.; Lunghi, B.; Fineschi, S.; Mirone, V.; d’Emmanuele di Villa Bianca, R.; Longo, N.I.C.; De Palma, R.; Sorrentino, R.; Lungarella, G.; Cirino, G.I. A new mouse model of Peyronie’s disease: An increased expression of hypoxia-inducible factor-1 target genes during the development of penile changes. Int. J. Biochem. Cell Biol. 2008, 40, 2638–2648.
- Layton, T.B.; Williams, L.; Yang, N.; Zhang, M.; Lee, C.; Feldmann, M.; Trujillo, G.; Furniss, D.; Nanchahal, J. A vasculature niche orchestrates stromal cell phenotype through PDGF signaling: Importance in human fibrotic disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2120336119.
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial-mesenchymal transition. J. Pathol. 2017, 241, 25–35.
- Abdelhakim, M.; Lin, X.; Ogawa, R. The Japanese experience with basic fibroblast growth factor in cutaneous wound management and scar prevention: A systematic review of clinical and biological Aspects. Dermatol. Ther. (Heidelb.) 2020, 10, 569–587.
- Mulhall, J.P.; Thom, J.; Lubrano, T.; Shankey, T.V. Basic fibroblast growth factor expression in Peyronie’s disease. Basic fibroblast growth factor expression in Peyronie’s disease. J. Urol. 2001, 165, 419–423.
- Burks, T.N.; Cohn, R.D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19.
- Lim, S.; McMahon, C.D.; Matthews, K.G.; Devlin, G.P.; Elston, M.S.; Conaglen, J.V. Absence of myostatin improves cardiac function following myocardial infarction. Heart Lung Circ. 2018, 27, 693–701.
- Cantini, L.P.; Ferrini, M.G.; Vernet, D.; Magee, T.R.; Qian, A.; Gelfand, R.A.; Rajfer, J.; Gonzalez-Cadavid, N.F. Profibrotic role of myostatin in Peyronie’s disease. J. Sex. Med. 2008, 5, 1607–1622.
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengellér, Z.K. AP39, a modulator of mitochondrial bioenergetics, reduces antiangiogenic response and oxidative stress in hypoxia-exposed trophoblasts: Relevance for preeclampsia pathogenesis. Am. J. Pathol. 2019, 189, 104–114.
- Li, X.; Xing, J.; Wang, H.; Yu, E. The SLC34A2-ROS-HIF-1-induced up-regulation of EZH2 expression promotes proliferation and chemo-resistance to apoptosis in colorectal cancer. Biosci. Rep. 2019, 39, BSR20180268.
- Kim, J.; Kim, B.; Kim, S.M.; Yang, C.E.; Song, S.Y.; Lee, W.J.; Lee, J.H. Hypoxia-induced epithelial-to-mesenchymal transition mediates fibroblast abnormalities via ERK activation in cutaneous wound healing. Int. J. Mol. Sci. 2019, 20, 2546.
- Cui, Y.; Wang, Y.; Men, C.; Wu, J.; Liu, L. Bioinformatics-based identification of potential hypoxia-related genes associated with Peyronie’s disease. Am. J. Mens Health 2022, 16, 15579883221111720.
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix. Biol. 2016, 49, 10–24.
- Randles, M.; Lennon, R. Applying proteomics to investigate extracellular matrix in health and disease. Curr. Top. Membr. 2015, 76, 171e196.
- Paulis, G.; De Giorgio, G.; Paulis, L. Role of oxidative stress in Peyronie’s disease: Biochemical evidence and experiences of treatment with antioxidants. Int. J. Mol. Sci. 2022, 23, 15969.
- Del Carlo, M.; Cole, A.A.; Levine, L.A. Differential calcium independent regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by interleukin-1beta and transforming growth factor-beta in Peyronie’s plaque fibroblasts. J. Urol. 2008, 179, 2447–2455.
- Watanabe, M.S.; Theodoro, T.R.; Coelho, N.L.; Mendes, A.; Leonel, M.L.P.; Mader, A.M.; Nader, H.B.; Glina, S.; Pinhal, M.A.S. Extracellular matrix alterations in the Peyronie’s disease. J. Adv. Res. 2017, 8, 455–461.
- Flevaris, P.; Vaughan, D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177.
- Davila, H.H.; Magee, T.R.; Zuniga, F.I.; Rajfer, J.; Gonzalez-Cadavid, N.F. Peyronie’s disease associated with increase in plasminogen activator inhibitor in fibrotic plaque. Urology 2005, 65, 645–648.
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmoulière, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 2012, 5 (Suppl. S1), S5.
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185.
- Szardening-Kirchner, C.; Konrad, L.; Hauck, E.W.; Haag, S.M.; Eickelberg, O.; Weidner, W. Upregulation of mRNA expression of MCP-1 by TGF-beta1 in fibroblast cells from Peyronie’s disease. World. J. Urol. 2009, 27, 123–130.
- Hatzimouratidis, K.; Eardley, I.; Giuliano, F.; Hatzichristou, D.; Moncada, I.; Salonia, A.; Vardi, Y.; Wespes, E.; European Association of Urology. EAU guidelines on penile curvature. Eur. Urol. 2012, 62, 543–552.
- Hao, M.; Han, X.; Yao, Z.; Zhang, H.; Zhao, M.; Peng, M.; Wang, K.; Shan, Q.; Sang, X.; Wu, X.; et al. The pathogenesis of organ fibrosis: Focus on necroptosis. Br. J. Pharmacol. 2022.
- Zorba, O.U.; Sirma, S.; Ozgon, G.; Salabas, E.; Ozbek, U.; Kadioglu, A. Comparison of apoptotic gene expression profiles between Peyronie’s disease plaque and tunica albuginea. Clin. Exp. Med. 2012, 21, 607–614.
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. Biomed. Res. Int. 2014, 616149.
- Loreto, C.; Barbagli, G.; Djinovic, R.; Vespasiani, G.; Carnazza, M.L.; Miano, R.; Musumeci, G.; Sansalone, S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and its death receptor (DR5) in Peyronie’s disease. A biomolecular study of apoptosis activation. J. Sex. Med. 2011, 8, 109–115.
- Hinz, B.; Lagares, D.N. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31.
- Garaffa, G.; Trost, L.W.; Serefoglu, E.C.; Ralph, D.; Hellstrom, W.J. Understanding the course of Peyronie’s disease. Int. J. Clin. Pract. 2013, 67, 781–788.
- Milenkovic, U.; Boeckx, B.; Lambrechts, D.; Janky, R.; Hatzichristodoulou, G.; van Renterghem, K.; Gevaert, T.; Cellek, S.; Bivalacqua, T.J.; De Ridder, D.; et al. Single-cell transcriptomics uncover a novel role of myeloid cells and T-lymphocytes in the fibrotic microenvironment in Peyronie’s disease. Eur. Urol. Focus 2022, 8, 814–828.
More
Information
Subjects:
Urology & Nephrology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
25 Jun 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No