Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexander V. Vershinin | -- | 2982 | 2023-06-23 15:26:29 | | | |
| 2 | Jessie Wu | -3 word(s) | 2979 | 2023-06-25 05:02:11 | | | | |
| 3 | Jessie Wu | Meta information modification | 2979 | 2023-06-26 10:12:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vershinin, A.V.; Elisafenko, E.A.; Evtushenko, E.V. Subtelomeric Heterochromatin in Rye. Encyclopedia. Available online: https://encyclopedia.pub/entry/45990 (accessed on 24 June 2026).
Vershinin AV, Elisafenko EA, Evtushenko EV. Subtelomeric Heterochromatin in Rye. Encyclopedia. Available at: https://encyclopedia.pub/entry/45990. Accessed June 24, 2026.
Vershinin, Alexander V., Evgeny A. Elisafenko, Elena V. Evtushenko. "Subtelomeric Heterochromatin in Rye" Encyclopedia, https://encyclopedia.pub/entry/45990 (accessed June 24, 2026).
Vershinin, A.V., Elisafenko, E.A., & Evtushenko, E.V. (2023, June 23). Subtelomeric Heterochromatin in Rye. In Encyclopedia. https://encyclopedia.pub/entry/45990
Vershinin, Alexander V., et al. "Subtelomeric Heterochromatin in Rye." Encyclopedia. Web. 23 June, 2023.
Copy Citation
The genome of cultivated rye, Secale cereale L., is considered one of the largest among species of the tribe Triticeae and thus it tops the average angiosperm genome and the genomes of its closest evolutionary neighbors, such as species of barley, Hordeum (by approximately 30–35%), and diploid wheat species, Triticum (approximately 25%).The review provides an analysis of the structural organization of subtelomeric heterochromatic regions of rye chromosomes with a description of the molecular mechanisms contributing to their size increase during evolution and the classes of DNA sequences involved in these processes.
tandem repeats
transposable elements
subtelomeric heterochromatin
rye
1. Tandem Repeats
In the three most widespread, well-studied and commercially valuable Triticeae species (wheat, rye and barley), rye is deemed to be the most promising for the analysis of and then for discussing the concept of genetic redundancy and considering the presence of selfish DNA in the genomes. The haploid genome of cultivated rye, Secale cereale L., (2n = 2x = 14) is about 8.0 Gbp/1C in size [1], being larger than the genome of an average angiosperm (5.6 Gbp) [2]. For comparison, the genomes of rye’s closest relatives, barley and wheat, the haploid chromosome number in them being the same as in any Triticeae species, 7, are 5.1–5.3 Gbp and 5.8–6.1 Gbp in size, respectively. Available whole genome assemblies with annotated genes of diploid species (Triticum urartu and Aegilops tauschii, ancestors of the cultivated wheat, Triticum aestivum), barley, Hordeum vulgare, and rye showed very close coding gene counts, about 40,000 [3][4][5][6][7]. Within the genus Secale, there is about a 15-percent variation in total genome size between the cultivated rye S. cereale (the largest) and the most ancient wild rye S. silvestre (the smallest) [1]. The differences in size between their respective genomes correlate with the differences in the sizes of the heterochromatic regions at the chromosome ends [8]. The facts as listed above obviously suggest that the cultivated rye’s superiority in genome size amongst its closest relatives is due to a higher abundance of various classes of repeated DNA sequences. This assumption is supported by the results of the first cereal DNA reassociation kinetic experiments, which showed that more than 90% of the rye genome consists of repetitive DNA [9].
Large heterochromatic blocks in subtelomeric regions represent a chromosomal feature of rye that its most closely related genera, wheat and barley, do not possess. As recently as at the end of the past century, these regions were analyzed for DNA composition. Some tandemly organized families were found to have extremely high monomer copy numbers as was indicated by strong in situ hybridization signals. All together, these families make up 8–12% of the entire rye genome [10][11]. The molecular structure, copy number and monomer lengths were furthermore determined for the three most abundant of them, pSc119.2, pSc200 and pSc250 [12][13]. They are made up of monomeric units 118 and 379, and are 571 bp in length, respectively, with pSc200 contributing to ~2.5% of the genome and pSc250 and pSc119.2 each contributing to ~1%. Fluorescence in situ hybridization (FISH) experiments suggest that the pSc200 and pSc250 blocks coincide close to the telomere, while some pSc119.2 copies are confined to interstitial sites. The pSc119.2 sequence is also present in some other cereals, but pSc200 and pSc250 are largely rye-specific.
As far as shorter tandem repeats (minisatellites) are concerned, the most probable mechanism promoting the emergence of two or a few successive DNA monomers is the duplication event that followed replication slippage [14]. It is possible that the same mechanism applies to longer monomers, with sizes close to those of tandem families in the rye genome. By sequencing an extended DNA region with BAC119C15 in it [15], researchers revealed a consistent pattern of the alteration of pSc200 monomers having an average of 93% homology, suggesting an initial duplication, the divergence of the primary sequences of the monomers within the dimer and the subsequent amplification of the dimers. It is likely that virtually any DNA sequence can serve as the initial monomer; several cases of tandem DNAs emerging from pieces of transposable elements have been described [16][17][18]. Dimer amplification and the preservation of tandem repeat arrays can take place in the course of replication or mitotic/meiotic recombination by means of multiple recombination events, such as unequal crossing over between sister chromatids, sequence conversion, translocations that exchange material between non-homologous chromosomes and transpositions [19][20][21]. FISH on meiotic chromosomes shows that each of the families, pSc200 and pSc250, is present on the arms of each chromosome as a separate domain, the size and varying stain intensity of which implies the presence of more than one array within each domain. This assumption is supported by blot-hybridized patterns after pulsed-field gel electrophoresis (PFGE) of DNA in BAC clones [15]. The maximum size of the pSc200 and pSc250 arrays is 550–600 kb [22]; however, the majority of the arrays in each rye chromosome are rather short, not longer than 80–100 kb.
The chromosomal domains consisting of pSc200 and pSc250 arrays are normally separated by non-tandem DNA sequences. Restriction analysis of DNA of the BAC clones in the BAC library of the short arm of rye chromosome 1 (1RS) followed by blot-hybridization showed that the pSc200 and pSc250 arrays, as with human alpha satellite DNA, develop HORs (higher order repeats), each consisting of two to eight monomers. A single HOR is longer than 3 kb (379 bp × 8) in pSc200 and is nearly 3.5 kb (571 bp × 6) in pSc250. The order of HORs and the ratio of different HORs are specific for each array within a single arm [15]. This implies that multiple recombination events had been taking place independently in different arrays of the same chromosome arm, leading to the emergence of HORs.
The tandem families pSc200 and pSc250 have different evolutionary histories. Some pSc200 copies have been found in hexaploid wheat, other Triticeae species [23][24] and more distantly related cereals, such as rice and oats. It gives grounds to assume that this family emerged about 45 MYA, when the rice and oat lineages split [25]. The pSc250 family is younger; its copies, very low in number, are found only in the Triticeae species [24], suggesting that its age is about 15 Myr. However, when scrutinizing the evolutionary history of the pSc200 and pSc250 families, one should remember that the amplification (expansion) of these families was not observed before the radiation of the genus Secale (1.7 MYA) and was running especially high in the cultivated rye (S. cereale) [8]. Thus, pSc200 and pSc250 did not take as long to expand as, for example, human alpha satellite DNA did, which is confirmed by high (not less than 90%) homology between the monomers within each of these families [15]. The evolutionary histories of these two families are strikingly different from that of the third high copy number family of tandem repeats, pSc119.2. This family is much more common across the tribe Triticeae than pSc200 and pSc250 are. Not only rye, but also various Triticum and Aegilops species, as well as most wild barley species [26] including Hordeum bulbosum, which is closely related to the cultivated barley, contain this family in a high copy number. A large number of variant monomers with different lengths and highly heterogeneous primary structures are an indication that sequence homogenization has never taken place in pSc119.2 [26]. By far the most surprising pSc119.2-related observation is that both H. vulgare ssp. spontaneum, which is an immediate ancestor of the cultivated barley (H. vulgare), and the cultivated barley itself are devoid of this family [27]. Thus, the beginning of the barley domestication process caused the mass deletion of thousands copies of pSc119.2 genome-wide in the predecessor species.
2. Transposable Elements
Transposable elements (TEs) are the predominant component of such large genomes as the cereal ones [28][29]. For this reason, researchers were not surprised to see that the sequencing of the DNA around the arrays of the pSc200 and pSc250 family tandems in BAC clones revealed various classes of these elements, basically LTR-containing retrotransposons. Are these two main classes of repeats in heterochromatic regions related in some way? Is the composition of the DNA surrounding the arrays of the tandem repeats specific to them—or is it unrelated to them? To answer these questions, researchers used 454 reads, which served as a basis for the rye genomic library [30]. The benefit of this approach is that no contigs are needed, which, if they were, would represent a major problem in duly profiling repeats, as the genome crawds with them. Nevertheless, the average length of the 454 reads is sufficient for spotting junction regions between tandem monomers and the DNA that surrounds them. Three sets of 454 sequencing-based reads were produced (Figure 1): two consisted of reads, which were essentially DNA in the “non-tandem DNA—pSc200 (or pSc250) tandem” junction regions, and the third sample consisted of reads with DNA residing in the rest of the genome [15]. Researchers' interest was to find out whether there was a correspondence between the copy number of separate TE families in the “non-tandem DNA-tandem DNA” junction regions and their copy number throughout the entire rye genome. The comparison was carried out using a database of eukaryotic repetitive elements, Repbase and the Triticeae Repeat Sequence Database (TREP). Researchers' analysis of the genome composition for rye showed that 73% of all TEs identified belonged to the gypsy-like superfamily of LTR-containing retrotransposons. Of all the TE families identified, the most abundant was the gypsy-like family Sabrina (Figure 2), which makes up about 15.5% of the total number of TEs identified genome-wide, which greatly exceeds the abundance of the next two top TE families, the CACTA superfamily and the gypsy-like family, WHAM. At the same time, the abundance of Sabrina dropped abruptly when this family was flanked by arrays of tandem repeats. Some TE families, common in the rye genome, virtually do not occur flanked by arrays of tandem repeats. For the example, neither CACTA nor Cereba nor Derami nor Sumana were flanked by pSc250, and Fatimawas never flanked by pSc200.
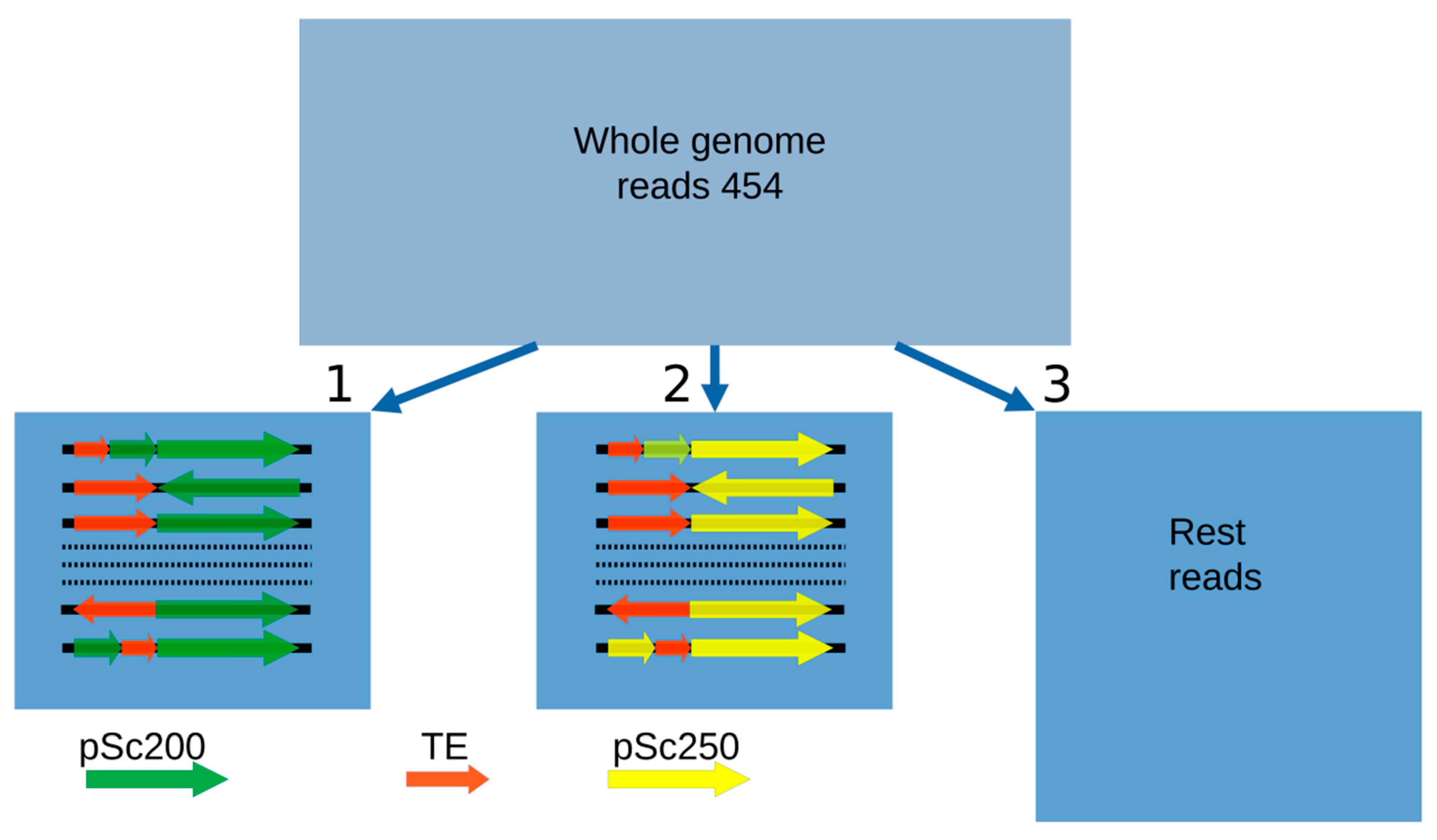
Figure 1. Three sets of 454 reads used for profiling TE families: (1) in “non-tandem DNA-pSc200 tandem” junction regions, (2) in the “non-tandem DNA—pSc250 tandem” junction regions and (3) in the rest of the rye genome. By way of illustration, Rectangles 1 and 2 display different possible variants of the reads.
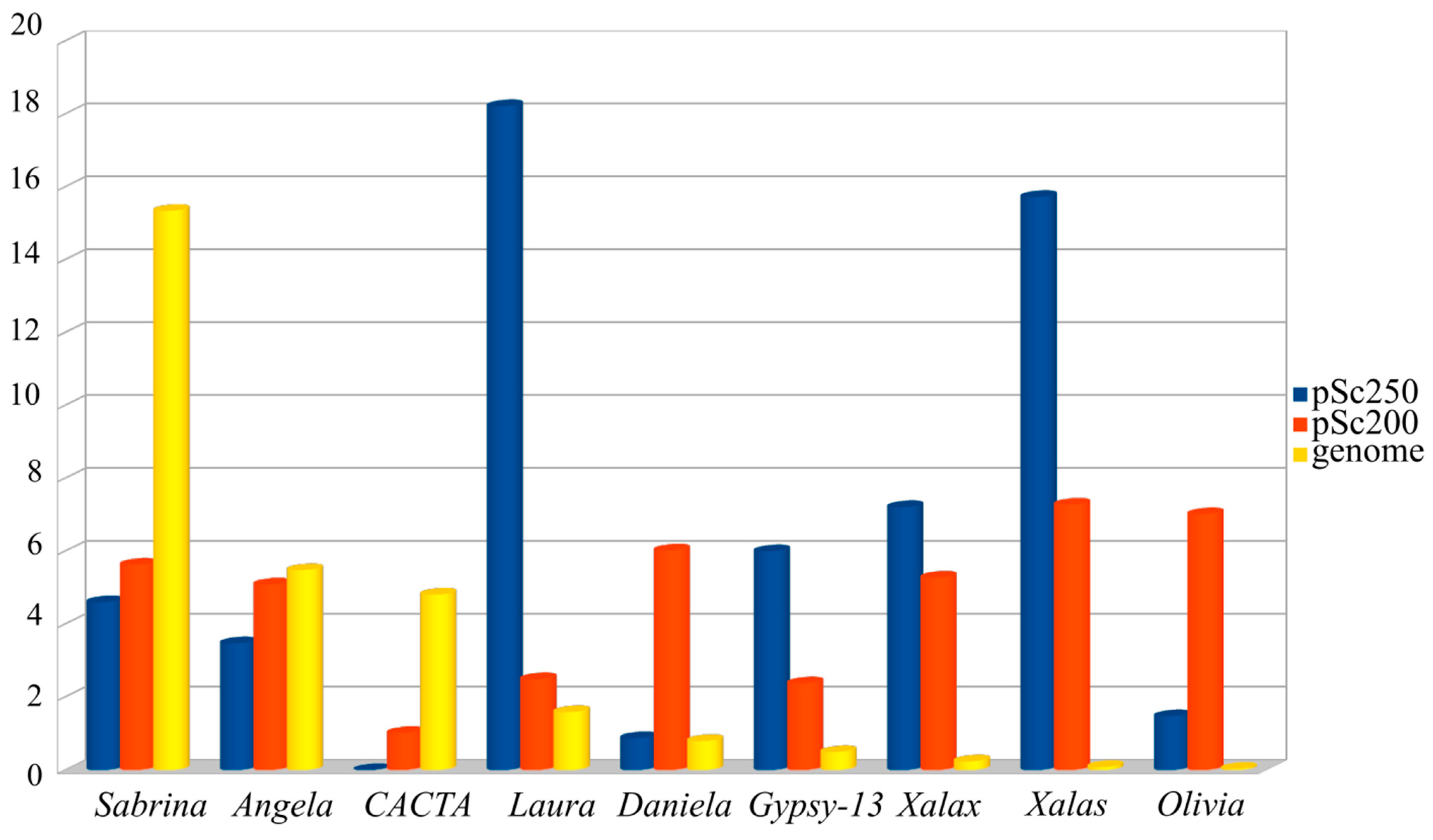
Figure 2. Presence of the most well-represented TE families in the rye genome (Sabrina, Angela, CACTA, yellow bars) and in the reads containing regions of junctions between non-tandem DNA and the monomers of pSc200 tandem repeats (Olivia, Daniela, Xalas, Xalax, red bars) and pSc250 tandem repeats (Laura, Xalas, Xalax, Gypsy-13_TA-I, blue bars).
Thus, a surprising feature revealed by comparing the abundance of the DNA sequences in the entire rye genome and when these sequences were flanked by the arrays of the tandem monomers pSc200 and pSc250 was a nearly reciprocal replacement of TE families. In terms of genome-wide abundance, Sabrina, CACTA and WHAM yield precedence to Daniela and Olivia flanked by pSc200 and Laura and Gypsy-13_TA-I flanked by pSc250, but the abundance of solo-LTRs designated as Xalas or Xalax is just rocketing, especially when these are flanked by pSc250. As the ectopic exchange model suggests [31], this should be indicative of very frequent ectopic exchanges in the immediate neighborhood of the tandem arrays pSc200 and pSc250 while these arrays were in the making. The main sources of solo-LTRs are probably unequal cross-overs and the within-chromosome ectopic recombination between the LTRs of the same or different elements, provided that they share quite extended homologous regions, such as those shared by Xalas and Xalax, and Daniela and Olivia. Vicient et al. [32] distinguish four forms of retrotransposon recombination. One of these forms is the LTR-LTR recombination, which results in solo-LTRs or tandem arrays consisting of LTRs and internal domains. According to this mechanism, if an LTR borders with any other DNA sequence, for example, pSc200 or pSc250 monomers, then the LTR-LTR recombination will generate a tandem array of these monomers next to the LTR.
While the mechanisms of recombination processes occurring within the arrays of tandem monomers and leading to the formation and genome-wide distribution of higher-order repeat units, varying in length and internal organization, have been known for decades, little is known about a potential impact that the immediate DNA neighborhood of the tandem monomer arrays might have on these processes. Researchers' analysis of the junction regions between TEs and the monomers of the tandem arrays pSc200 and pSc250 revealed that most TE transpositions occurred either directly to the monomers of the tandem array or next to them following ligation, which added a very short spacer, DNA 1–10 bp in length—a situation typical of non-homologous end joining (NHEJ). About 90% of these junction regions put together pSc250 and two groups, Laura and Xalas, the most abundant when surrounded by TEs: about 70% for pSc200 and Daniela, and 58% for pSc200 and Olivia.
In many organisms, recombination events occur at certain sites, the so-called “hot spots”, which have a particular nucleotide context [33]. In the nucleotide context of DNA of the transposable elements bordering the tandem arrays pSc200 and pSc250, motifs that could participate in recombination other than HR (homologous recombination) or NHEJ (non-homologous end-joining) and promote the propagation of the arrays of tandem monomers were revealed to occur with a probability much higher than could have been expected by random chance [15]. The lengths of these motifs are 8-12 bp, which are sufficient for recombinases to start aligning a single-strand DNA with a homologous duplex elsewhere in the genome, which can promote recombination. Thus, the LTRs containing microhomologous DNA within this TE inserted next to the pSc200 or pSc250 monomers could, together with captured monomers, recombine these sites with other copies of the same TE occurring elsewhere in the genome and thus promote the distribution of the tandem monomer arrays.
3. Rye Genomic Libraries Detailed the Organization of Subtelomeric Heterochromatin
The rye genomic libraries generated in recent years using modern DNA sequencing techniques and contig assemblies [6][7] allow for large genomic regions to be analyzed. As is known, the heterochromatic chromosome regions and, first of all, long arrays of tandemly organized monomers are especially difficult to read. Nevertheless, the contigs in the genome libraries mentioned are long enough to shed light on the structural organization of the subtelomeric heterochromatic regions of the rye chromosomes, including both classes of repeats: monomer tandems and copies of TE families. Some contigs begin from the chromosome ends, as the presence of the contig region containing telomeric monomers (TTTAGGG)n suggests. The telomere monomers are immediately followed by the array of the pSc119.2 monomers on one arm of chromosome 3R [7] and the pSc200 array on the other [6]. However, the arrays of telomeric and subtelomeric repeats are not immediate neighbors in all chromosomes. In the rye line ‘Weining’, the chromosome 7R telomere and the array of pSc250 monomers are separated by four pieces of the transposable elements Xalax and Gypsy13_TA-I, and the pSc250 array itself is interspersed with pieces of various transposable elements. The arrangement of the arrays of all of these tandem repeat families, pSc200, pSc250 and pSc119.2, being relative to each other may be extremely varying; they may (1) lie very close to each other, (2) be separated by variously sized tracks consisting mostly of pieces of diverse TE families or (3) occur in different combinations.
The schematic in Figure 3 shows one of the longest scaffolds, s291 (line ‘Lo7’), as an example to illustrate the structural organization of a chromosome region and to list all of the above-described main characteristics of subtelomeric heterochromatin. The pSc200 cluster in it is 258.3 kb in size and consists of several long monomer arrays interrupted by three regions. One of these regions, which is about 17 kb in length and located at the beginning of the scaffold, is populated by several rearranged copies of the copia-like family WIS-2, while the other two, being rather short, contain some of the WIS-2 sequences and some of the gypsy-like family Olivia. From position 80 kb, the array of pSc200 monomers forms an 1897-bp-long region consisting of 13 HORs (purple in the schematic), each consisting of five monomers.
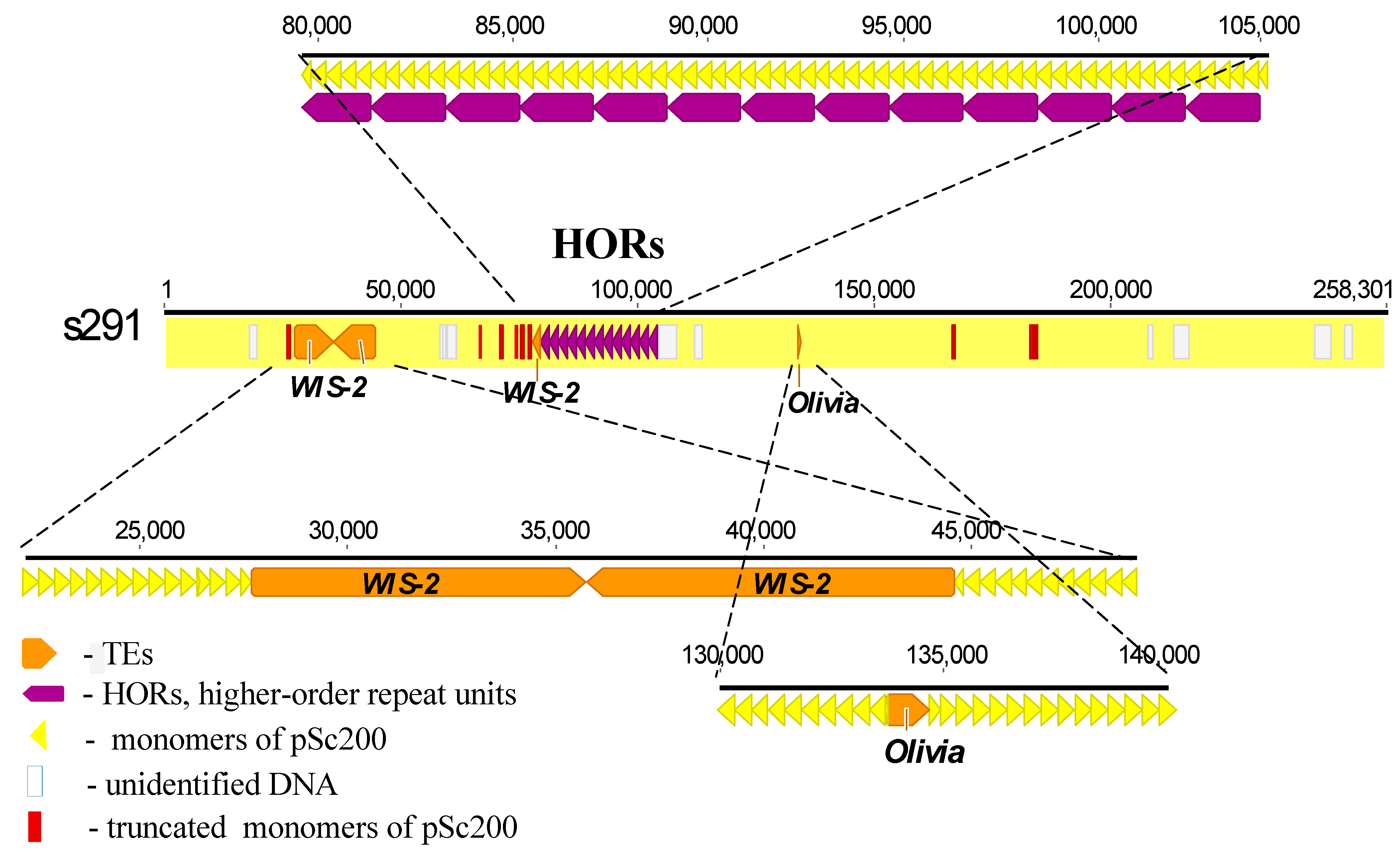
Figure 3. Schematic of the molecular organization of heterochromatin in rye subtelomeres (line ‘Lo7’), with the scaffold s291 as an example. pSc200 monomers (yellow finger-post arrows) and transposable elements (brown finger-post arrows) alternate with pSc200 monomers. Thirteen HOR units, each consisting of five pSc200 monomers, are pointed to by purple arrows. Truncated pSc200 monomers (red rectangles) are scattered along the scaffold with increased concentration near the 5’-ends of the HORs and the truncated copy of WIS-2. The numbers at the top show distances in kilobases.
It is possible that more improved sequencing techniques and the assembly of genomic libraries will allow researchers to reveal genes within the subtelomeric heterochromatin of the rye chromosomes. Some genes have been found and described within small regions of constitutive heterochromatin in Drosophila melanogaster [34]; however, these regions do not have powerful arrays of tandem repeats in them, where as such regions in rye chromosomes do. The chicken pan-genome constructed from 20 de novo assembled genomes with high sequencing depth [35] brings hope that functional genes within the subtelomeric heterochromatic regions of rye chromosomes will eventually be found. As a result, the authors found 1335 previously unannotated protein-coding genes, the majority of which were located in subtelomeric chromosome and minichromosome regions and were surrounded by huge arrays of tandem repeats that made sequencing impossible. Looking over the amount of knowledge that researchers currently have about the structural organization of subtelomeric heterochromatin in rye chromosomes, researchers must confess that it is difficult to make assumptions about and understand how the long arrays of tandem DNA repeats that are prevalent in it and occur genome-wide in thousands of copies, in which ever-running recombination processes affect the neighboring TEs, most of which appear in the form of relatively short pieces such as solo-LTRs, could participate in encoding, regulation or any other molecular process related to survival, reproduction or behavior, or any other process that is beneficial to the metabolism and well-being of living organisms. It is therefore logical and reasonable to assume that what DNA in such chromosome regions does is only ensure its survival, which is consistent with the criteria of “selfish DNA” and the concept of genetic redundancy in eukaryotic genomes.
References
- Leitch, I.J.; Johnston, E.; Pellicer, J.; Hidalgo, O.; Bennett, M.D. Plant DNA C-Values Database (Release 7.1). 2019. Available online: https://cvalues.science.kew.org/ (accessed on 12 September 2022).
- Rabinowicz, P.D.; Bennetzen, J.L. The maize genome as a model for efficient sequence analysis of large plant genomes. Curr. Opin. Plant Biol. 2006, 9, 149–156.
- Luo, M.-C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; McGuire, P.E.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502.
- Ling, H.Q.; Ma, B.; Shi, X.; Liu, H.; Dong, L.; Sun, H.; Cao, Y.; Gao, Q.; Zheng, S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428.
- Jayakodi, M.; Padmarasu, S.; Haberer, G.; Bonthala, V.; Gundlach, H.; Monat, C.; Lux, T.; Kamal, N.; Lang, D.; Himmelbach, A.; et al. The barley pan-genome reveals the hidden legacy of mutation breeding. Nature 2020, 588, 284–289.
- Rabanus-Wallace, M.T.; Hackauf, B.; Mascher, M.; Lux, T.; Wicker, T.; Gundlach, H.; Baez, M.; Houben, A.; Mayer, K.F.X.; Guo, L.; et al. Chromosome-scale genome assembly provides insights into rye biology, evolution and agronomic potential. Nat. Genet. 2021, 53, 564–573.
- Li, G.; Wang, L.; Yang, J.; He, H.; Jin, H.; Li, X.; Ren, T.; Ren, Z.; Li, F.; Han, X.; et al. A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat. Genet. 2021, 53, 574–584.
- Cuadrado, A.; Jouve, N. Evolutionary trends of different repetitive DNA sequences during speciation in the genus Secale. J. Hered. 2002, 93, 339–345.
- Flavell, R.B.; Bennett, M.D.; Smith, J.B.; Smith, D.B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 1974, 12, 257–269.
- Bedbrook, J.R.; Jones, J.; O’Dell, M.; Tompson, R.; Flavell, R. A molecular description of telomeric heterochromatin in Secale species. Cell 1980, 19, 545–560.
- Jones, J.D.G.; Flavell, R.B. The mapping of highly-repeated DNA families and their relationship to C-bands in chromosomes of Secale cereale. Chromosoma 1982, 86, 595–612.
- McIntyre, C.L.; Pereira, S.; Moran, L.B.; Appels, R. New Secale cereale (rye) DNA derivatives for the detection of rye chromosome segments in wheat. Genome 1990, 33, 317–323.
- Vershinin, A.V.; Schwarzacher, T.; Heslop-Harrison, J.S. The large-scale organization of repetitive DNA families at the telomeres of rye chromosomes. Plant Cell 1995, 7, 1823–1833.
- Ames, D.; Murphy, N.; Helentjaris, T.; Sun, N.; Chandler, V. Comparative analyses of human single- and multilocus tandem repeats. Genetics 2008, 7, 603–613.
- Evtushenko, E.V.; Levitsky, V.G.; Elisafenko, E.A.; Gunbin, K.V.; Belousov, A.I.; Šafář, J.; Doležel, J.; Vershinin, A.V. The expansion of heterochromatin blocks in rye reflects the co-amplification of tandem repeats and adjacent transposable elements. BMC Genom. 2016, 17, 337.
- Tek, A.L.; Song, J.; Macas, J.; Jiang, J. Sobo, a recently amplified satellite repeat of potato, and its amplifications for the origin of tandemly repeated sequences. Genetics 2005, 170, 1231–1238.
- Sharma, A.; Wolfgruber, T.K.; Presting, G.G. Tandem repeats derived from centromeric retrotransposons. BMC Genom. 2013, 14, 142.
- Vondrak, T.; Robledillo, L.A.; Novak, P.; Koblizkova, A.; Neumann, P.; Macas, J. Characterization of repeat arrays in ulyra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J. 2020, 101, 484–500.
- Smith, G.P. Evolution of repeated DNA sequences by unequal crossover. Science 1976, 191, 528–535.
- Dover, G. Molecular drive. A cohesive mode of species evolution. Nature 1982, 299, 111–117.
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220.
- Alkhimova, O.G.; Mazurok, N.A.; Potapova, T.A.; Zakian, S.M.; Heslop-Harrison, J.S.; Vershinin, A.V. Diverse patterns of the tandem repeats organization in rye. Chromosoma 2004, 113, 42–52.
- Xin, Z.-Y.; Appels, R. Occurrence of rye (Secale cereale) 350-family DNA sequences in Agropyron and other Triticeae. Plant Syst. Evol. 1987, 160, 65–76.
- Vershinin, A.V.; Alkhimova, E.G.; Heslop-Harrison, J.S. Molecular diversification of tandemly organized DNA sequences and heterochromatic chromosome regions in some Triticeae species. Chromosome Res. 1996, 4, 517–525.
- Gaut, B.S. Evolutionary dynamics of grass genomes. New Phytol. 2002, 154, 15–28.
- Contento, A.; Heslop-Harrison, J.S.; Schwarzacher, T. Diversity of a major repetitive DNA sequence in diploid and polyploidy Triticeae. Cytogenet. Genome Res. 2005, 109, 34–42.
- Taketa, S.; Ando, H.; Takeda, K.; Harrison, G.E.; Heslop-Harrison, J.S. The distribution, organization and evolution of two abundant and widespread repetitive DNA sequences in the genus Hordeum. Theor. Appl. Genet. 2000, 100, 169–176.
- Charles, M.; Belcram, H.; Just, J.; Huneau, C.; Viollet, A.; Couloux, A.; Segurens, B.; Carter, M.; Huteau, V.; Coriton, O.; et al. Dynamics and differential proliferation of transposable elements during the evolution of the B and A genomes of wheat. Genetics 2008, 180, 1071–1086.
- Wicker, T.; Taudient, S.; Houben, A.; Keller, B.; Graner, A.; Platzer, M.; Stein, N. A whole-genome snapshot of 454 sequences exposes the composition of the barley genome and provide evidence for parallel evolution of genome size in wheat and barley. Plant J. 2009, 59, 712–722.
- Martis, M.M.; Zhou, R.; Haseneyer, G.; Schmutzer, T.; Vrana, J.; Kubalakova, M.; Konig, S.; Kugler, K.G.; Scholz, U.; Hackauf, B.; et al. Reticulate evolution of the rye genome. Plant Cell 2013, 25, 3685–3698.
- Peterson-Burch, B.D.; Nettleton, D.; Voytas, D.F. Genomic neighbourhoods for Arabidopsis retrotransposons: A role for targeted integration in the distribution of the Metaviridae. Genome Biol. 2004, 5, R78.
- Vicient, C.M.; Kalendar, R.; Schulman, A.H. Variability, recombination, and mosaic evolution of the barley BARE-1 retrotransposon. J. Mol. Evol. 2005, 61, 275–291.
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008, 40, 1124–1129.
- Devlin, H.H.; Bingham, B.; Wakimoto, B.T. The organization and expression of the light gene, a heterochromatic gene of Drosophila melanogaster. Genetics 1990, 125, 129–140.
- Li, M.; Sun, C.; Xu, N.; Bian, P.; Tian, X.; Wang, X.; Wang, Y.; Jia, X.; Heller, R.; Wang, M.; et al. De novo assembly of 20 chicken genomes reveals the undetectable phenomenon for thousands of core genes on microchromosomes and subtelomeric regions. Mol. Biol. Evol. 2022, 39, msac066.
More
Information
Subjects:
Anatomy & Morphology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
686
Revisions:
3 times
(View History)
Update Date:
26 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No