Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karolina Henryka Czarnecka-Chrebelska | -- | 2579 | 2023-06-20 17:00:21 | | | |
| 2 | Sirius Huang | Meta information modification | 2579 | 2023-06-21 04:08:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Czarnecka-Chrebelska, K.H.; Mukherjee, D.; Maryanchik, S.V.; Rudzinska-Radecka, M. Biological and Genetic Mechanisms of COPD. Encyclopedia. Available online: https://encyclopedia.pub/entry/45877 (accessed on 11 June 2026).
Czarnecka-Chrebelska KH, Mukherjee D, Maryanchik SV, Rudzinska-Radecka M. Biological and Genetic Mechanisms of COPD. Encyclopedia. Available at: https://encyclopedia.pub/entry/45877. Accessed June 11, 2026.
Czarnecka-Chrebelska, Karolina H., Debjita Mukherjee, Sofya V. Maryanchik, Magdalena Rudzinska-Radecka. "Biological and Genetic Mechanisms of COPD" Encyclopedia, https://encyclopedia.pub/entry/45877 (accessed June 11, 2026).
Czarnecka-Chrebelska, K.H., Mukherjee, D., Maryanchik, S.V., & Rudzinska-Radecka, M. (2023, June 20). Biological and Genetic Mechanisms of COPD. In Encyclopedia. https://encyclopedia.pub/entry/45877
Czarnecka-Chrebelska, Karolina H., et al. "Biological and Genetic Mechanisms of COPD." Encyclopedia. Web. 20 June, 2023.
Copy Citation
Chronic obstructive pulmonary disease (COPD) is one of the most prevalent chronic adult diseases, with significant worldwide morbidity and mortality. Although long-term tobacco smoking is a critical risk factor for this global health problem, its molecular mechanisms remain unclear. Several phenomena are thought to be involved in the evolution of emphysema, including airway inflammation, proteinase/anti-proteinase imbalance, oxidative stress, and genetic/epigenetic modifications. Furthermore, COPD is one main risk for lung cancer (LC), the deadliest form of human tumor; formation and chronic inflammation accompanying COPD can be a potential driver of malignancy maturation.
chronic obstructive pulmonary disease
lung cancer
biomarkers
smokers
emphysema
1. Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by lung airflow limitation and tissue destruction; it is the third leading cause of death worldwide [1]. COPD can result from chronic bronchitis with characteristic airway inflammation and scarring [2]. Tobacco smoking is the most common COPD risk factor. However, many other inhaled irritants are still involved (burning biomass fuels, smoke, air pollutants, and chemicals), leading to heterogeneous COPD phenotypes [3][4].
2. COPD Causes
Chronic obstructive pulmonary disease (COPD) involves the chronic inflammatory condition of the lung, particularly the conducting airways and parenchyma [5]. Imbalances accompany the process of progressive inflammation in proteinase/anti-proteinase activity or oxidant–antioxidant balance, triggering emphysema formation (the abnormal enlargement of the air spaces located peripherally from the terminal bronchioles and destruction of the walls of these structures) [1][2][3]. Emphysema may lead to further changes in lung tissue, i.e., deterioration of elasticity, poor expiratory flow, gas trapping, and impairment of the gas exchange [2].
COPD affects millions worldwide, making it a significant health burden connected with high healthcare costs [6]. COPD is attributed to increasing morbidity and mortality in low- and middle-income countries, especially in acute exacerbation patients [7]. The impact of the COPD issue is that many people are underdiagnosed, and only 50% of patients are adequately treated with medications [8].
Tobacco smoking is the most common risk factor for developing COPD, with patients more likely to develop the disease if they smoked one pack per day for 20 years or more [3]. However, the absolute risk of COPD developing in continuous smokers is around 25%, suggesting that there may be other predisposing factors, such as genetic, epigenetic, or host-dependent factors (Figure 1).
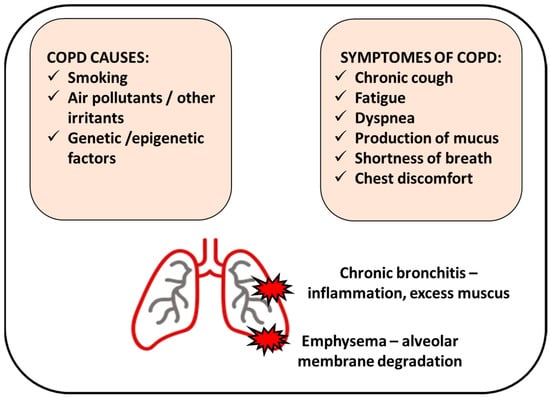
Figure 1. Chronic obstructive pulmonary disease (COPD) manifests by inflamed airways and damaged lung tissue. Smoking cigarettes is the most common cause of COPD; however, other factors can be involved. COPD includes chronic bronchitis (inflammation of the bronchial tubes that causes a persistent cough) and emphysema (damage of the air sacs).
Among these factors are gene polymorphisms in molecules, such as α1-anti-trypsin [9], tumor necrosis factor (TNF-α) [10], matrix metalloproteinases (MMPs) [11], or antioxidant gene dependence [12], which show race, age, and disease severity/phenotype dependence [13][14][15].
Next, the disease can be developed from inhalation of smoke from burning biomass fuels, exposure to pollutants and chemicals, or other inhaled irritants [4].
The characteristic COPD symptoms are dyspnea, forced expiratory volume in 1 s (FEV1), cough, and sputum production; less common symptoms are wheezing, tightness, and chest congestion [16]. However, reported symptoms showed seasonal and weekly variability and differed depending on the patient population’s disease severity. Frequent exacerbations in COPD patients may eventually result in increased airway inflammation with higher levels of eosinophils and neutrophils and enhanced inflammatory mediators such as cytokines [17].
All cigarette smokers have some lung inflammation, but those who develop COPD present an enhanced or abnormal response to inhaling toxic agents [18]. This amplified response may result in mucous hypersecretion (chronic bronchitis), tissue destruction (emphysema), and disruption of regular repair and defense mechanisms, causing slight minor inflammation and fibrosis (bronchiolitis) [18] (Figure 1).
Two major pathologic processes cause the progressive airflow limitation in COPD: remodeling and narrowing of small airways and destruction of the lung parenchyma with consequent destruction of the alveolar attachments of these airways as a result of emphysema [19] (Figure 1).
After smoking cessation, the symptoms usually decrease. However, lung tissue transformation and lung function are not restored, and increased airway resistance persists. Furthermore, the inflammation persists, contributing to the irreversibility of the decreased lung function. This can be associated with pathological tissue-like fibrosis and inflammation that reduces the diameter of the airway lumen [20].
Due to the overlapping deterioration of processes controlling lung physiology, ageing is regarded as an independent COPD risk factor. The age-related aspects are (i) the decline in the strength of the respiratory muscles due to cardiac function, (ii) age-related reduction in peripheral muscle mass (related to decreased physical activity and nutritional status), and (iii) the geometric changes in the rib cage [21]. Moreover, due to the progressive dilatation of the alveolar ducts and loss of supporting tissues for the peripheral airways, the static elastic recoil of the lung diminishes. Thus, the ageing population is more vulnerable to COPD due to physiological changes in the lung. Since life expectancy has risen, and about 20% of people in developed countries are over 65 years old, focusing on the process leading to lung dysfunction is highly important.
3. Genetics of COPD
3.1. Gene Polymorphisms
Genome-wide association studies have been conducted to find the genes responsible for the onset or progression of COPD [22] (Table 1). One detected gene directly responsible for COPD appearance is alpha-1-antitrypsin (AAT) [23]. AAT deficiency, due to alterations in DNA sequence, is associated with disease development in 1–2% of the affected population. Occurrence of specific alleles, ATT*Z allele homozygosity (Pi Z) or heterozygosity of the Z allele with a null allele, is related with AAT deficiency. In Pi Z homozygotes, the AAT protein polymerize reduces the amount of protein circulating in the body and causes a decreased serum level of AAT [24]. The accumulation of AAT in the hepatocytes leads to liver disorders, such as cirrhosis, hepatitis, and cancer [25]. Since AAT acts as a plasma protease inhibitor of the enzyme leukocyte elastase (present in neutrophils), AAT deficiency leads to loss of the natural defense mechanisms due to lack of proper protease activities and results in inflammation that triggers emphysema, a common condition observed in COPD patients [24][26].
Significant genes connected with susceptibility towards COPD showed a nucleotide polymorphism (SNP) pattern with point mutations causing the replacement of a nucleotide with another in a particular gene locus, resulting in different alleles [27]. In a case-control cohort study conducted by Pillai et al. (2009), two SNPs were observed at the α-nicotinic acetylcholine receptor (CHRNA3/5 in chromosome 5) locus to be significant in lung dysfunction and increasing the risk for COPD (12.2% cases in the population presented this gene modification) [28]. Extensive cohort investigations of 1633–3000 individuals involving controls (smokers) and COPD patients were studied for SNPs and their pedigree analysis. The two SNPs (rs8034191 and rs1051730) in the CHRNA3/5 locus were found to be the most reliably associated with COPD and significantly associated with lung function or the FEV1 parameter [29]. Furthermore, the observation provided by Wilk et al., 2012, showed that CHRNA3/5 is a risk factor independent of smoking [30].
Next, the HHIP (Hedgehog interacting protein) locus in chromosome 4q31, a part of the hedgehog gene family, was involved in morphogenesis and lung development [28][29][30].
In the same region on chromosome 15q25.1, the IREB2 (Iron responsive element binding protein 2) and ADPHD1 (Aspartate beta-hydroxylase domain containing 1) were found to be involved in COPD development. The HTR4 (5-hydroxytryptamine receptor 4) gene was also responsible for FEV1/FVC changes. Another important locus was identified on chromosome 19q13, where the CYP2A6 (Cytochrome P450 family 2 subfamily A member 6) gene was significant in smoking populations [31][32][33]. The CYP2A6 gene controls the enzyme required for nicotine metabolism and is vital in smokers. A study by Bloom and colleagues also implicated the gene responsible for hypoxia, EGLN2 (Egl-9 family hypoxia inducible factor 2), playing a role in COPD; furthermore, CYP2A6 acts independently of the nicotine metabolism and hence can be responsible for COPD in both smokers and non-smokers [34]. Other genome-wide association studies and meta-analysis examinations have also shown the identification of 39 new loci (such as EEFSEC, DSP, MTCL1, and SFTPD) relating COPD to lung function, asthma, pulmonary fibrosis, lung composition (cells, tissues, and smooth muscles) and other comorbidity factors [35][36].
Table 1. Gene polymorphisms in COPD.
| Gene Identified | Location of Polymorphisms | Critical Effects | Reference |
|---|---|---|---|
| α-1-antitrypsin (AAT) | ATT*Z allele (Pi Z) homozygosity, single amino acid substitution causing base pair changes | low levels of AAT in serum, accumulation in hepatocytes leading to liver damage, neutrophil inactivity, emphysema | [23][24][25][26] |
| Alpha-nicotinic acetylcholine receptor | 2 SNPs (rs8034191 and rs1051730) at locus of CHRNA3/5 in chromosome 5 | lung dysfunction (deviations in FEV1 parameter) | [28][29] |
| HHIP (Hedgehog interacting protein) | chromosome 4q31 (HHIP mutations) |
developmental problems in the lung and abnormality during morphogenesis | [28][29][30] |
| IREB2 (Iron responsive element binding protein 2) | chromosome 15q25.1 (SNP rs7937) |
lung developmental changes and emphysema | [31][37] |
| ADPHD1 (Aspartate beta-hydroxylase domain containing 1) | chromosome 15q25.1 | airflow obstruction, AAT deficiency | [31] |
| HTR4 (5-hydroxytryptamine receptor 4) | chromosome 5q31-q33 | FEV1/FVC changes, airflow obstruction | [33] |
| CYP2A6 (Cytochrome P450 family 2 subfamily A member 6) | chromosome 19q13 | nicotine metabolism affected | [31][34] |
| EGLN2 (Egl-9 family hypoxia inducible factor 2) | chromosome 19q13.2 | hypoxia response destroyed | [34] |
3.2. Epigenetic Regulation (Methylation and Deacetylation)
DNA methylation is a reversible modification of DNA structure involving the transfer of a methyl group onto the C5 position of the cytosine, often as part of a CpG island or cluster [38]. DNA methylation is found to play a critical role in COPD development, and this epigenetic mechanism can be altered by cigarette smoking (Table 2) [39]. Lung macrophages substantially affect the polarization of innate and adaptive immunity and the recognition and elimination of bacteria. In this context, it was detected that several inflammatory/immune-related genes of lung macrophages, including HSH2D (Hematopoietic SH2 domain containing), SNX10 (Sorting nexin 10), CLIP4 (CAP-Gly domain containing linker protein family member 4), and TYKZ are 95 CpG loci with significant difference of methylation [40]. As the authors confirmed, this DNA methylation of selected gene loci in lung macrophages is associated with metabolic differences regionally in the lung.
Next, mitochondrial transcription factor A (mtTFA) was remarkably decreased in the skeletal muscle of COPD patients, which was enhanced by cigarette smoke [41]. This phenomenon was positively correlated with the initiation and progression of COPD [41].
Interestingly, the methylation pattern changes can be affected by air pollution components like particulate matter (PM), ozone, nitrogen oxides, and polyaromatic hydrocarbons [42]. Twenty-seven differentially methylated regions (DMRs) in CpGs in NEGR1 (Neuronal growth regulator 1), ARID5A (AT-rich interaction domain 5 A), FOXl2 (Forkhead box 12), WDR46 (WD repeat domain 46), AKNA (AT-hook transcription factor), and SYTL2 (Synaptotagmin like 2) genes were correlated with prolonged exposure to PM10 and nitrogen dioxide [43].
Differentially methylated regions were detected in parenchymal fibroblasts in COPD, located in genes such as TMEM44 (Transmembrane protein 44), RPH3AL (Rabphilin 3 A like), WNT3A (Wnt family member 3 A), HLA-DP1 (Major histocompatibility complex, class II, DP beta 1), and HLA-DRB5 (Major histocompatibility complex, class II, DR beta 5) [44]. In addition, GWAS suggested that common SERPINA1 variants might influence COPD risk and associated lung function phenotypes [45]. Furthermore, hypermethylation of SERPINA1 in COPD patients was associated with tobacco addiction [46]. As a result, this epigenetic change can affect excessive mucus secretion and production, and goblet cell metaplasia can cause COPD.
Histone acetylation is a reversible epigenetic change unequivocally associated with increasing the propensity for gene transcription [47]. Histone deacetylation and histone acetylation comprise two enzyme families (histone acetyltransferases (HATs) and histone deacetylases (HDACs) [48] and play an influential role in the occurrence of inflammation in COPD [49]. Studies proved the changeability of the acetylation/deacetylation balance toward acetylation in patients with COPD (Table 2) and resultant inflammation [50]. An increase in acetylated histone 4 was found in current smokers; conversely, ex-smokers with COPD showed an increase in histone 3.
Cigarette smoking and oxidative stress are two significant features to inhibit inflammation in lung parenchyma and airways in COPD cases. Consequently, cigarette smoking elevated oxidant stress and promoted COPD glucocorticoid resistance during patients’ treatment. It was associated with higher HDAC2 activity [51]. Ding et al. reported Trichostatin A-an inhibitor of HDAC1/2-suppressing skeletal muscle atrophy and histomorphological alteration in COPD individuals [52]. H3K9 histone acetylation was high in the COPD-diseased human bronchial epithelial group [53].
Table 2. Gene methylation and histone acetylation in COPD.
| Epigenetic Mechanism | Altering Factors | Targets | Phenotype/Function in COPD Context | Reference |
|---|---|---|---|---|
| Methylation | cigarette smoking, air pollution |
HSH2D, SNX10, CLIP4, TYKZ | regulation of lung macrophage activity, maintaining lung metabolic balance | [40] |
| mtTFA | hypermethylation of the promoter is associated with the initiation and progression of COPD | [41] | ||
| NEGR1, ARID5A, FOXl2, WDR46, AKNA, SYTL2 | air pollution-dependent regulation of gene expression in Asians | [43] | ||
| HLX, SPON2 | alteration of functional gene expression in parenchymal fibroblasts | [54] | ||
| IREB2, PSMA4 | smoke-independent association of COPD with genetic variants in chromosome 15q25.1 | [55] | ||
| Acetylation | cigarette smoking, regulators of HDACs activity (Trichostatin A) |
Histones: H3K9, H3, H4 | increased levels are associated with inflammation, active gene transcription | [53] |
3.3. Transcriptional Regulation and Splicing
Transcriptional regulation (effective mechanism in proteostasis) in COPD is affected by malfunction of factors, including β-catenin, TGF-β1, and SMAD signaling. These molecular changes are accompanied by an epithelial to mesenchymal transition (EMT) [56] that can result in organ fibrosis or malignant tumorigenesis [57]. The transcription factor clusters of β-catenin/Snail1/Twist were upregulated, translocated in the nucleus in COPD patients, and correlated with EMT activity and airway obstruction [58]. Next, TGF-β1 was enhanced in COPD samples but was not found to be related to EMT or airflow obstruction. On the other hand, pSMAD was upregulated in the smoking COPD populations and directly associated with EMT and airflow obstruction. mRNA post-transcriptional alterations in COPD cases were closely related to oxidative stress and inflammation [59] and were mediated by factors like RNA-binding proteins (RBPs), microRNA (miRNA), and long non-coding RNA (lncRNA) and posttranscriptional modifications, such as polyadenylation, pre mRNA splicing, and mRNA editing and turnover [60].
Non-encoding RNAs that are not translated into proteins are mainly divided into two categories based on size: (i) short-chain non-coding RNAs (including miRNAs) and (ii) long non-coding RNAs (lncRNAs) [61]. Both short and long non-coding RNA structures were found as potential biomarkers in COPD prognostication and diagnosis. Qu et al. reported that the lncRNA ENST00000502883.1 was decreased in peripheral blood mononuclear cells in COPD patients. At the same time, Qie et al. found that lncRNAs, including ENST00000447867 and NR-026690, were significantly upregulated in CD4+ T cells of COPD samples [62].
Following, miR-1236 was found to bind to 3/-UTR of TLR4 mRNA and pose the risk of ventilator-associated pneumonia in COPD patients. miR-206 was upregulated in skeletal muscle and plasma of COPD patients and associated with advanced disease [63].
MiR-206 expression was higher in the lung tissues, and the manifestation of NOTCH3 and VEGFA mRNAs was decreased in the COPD group [64]. MiR-199a-5p was repressed by approximately four times and enhanced regulatory T cells in COPD patients compared to healthy smokers [65]. Interestingly, the miRs were differently regulated by tobacco smoking and biomass exposure; miR-34a was downregulated in COPD exposed to biomass compared with tobacco smokers [66].
Some of the RBPs, including iron-responsive element binding protein 2 (IREB2), Human antigen R (HuR), and T-cell antigen 1 (TIA-1), are highly expressed in the lungs of COPD patients. In IREB2 knockout mice, smoke-induced pulmonary inflammation was found, but mucociliary airway clearance was regular [67][68][69][70]. Next, the miR-218-5p in human bronchial epithelial cells upon cigarette smoke extract exposure leads to higher overexpression of chemokines, such as CCL20 and CXCL8, which play an essential role in COPD progression [71].
The genes associated with COPD progression differ from normal genes due to the transcriptional complexity of the gene loci that can produce up to 2.65 transcripts per gene— this is much more than the expected number of transcripts [72]. This phenomenon was connected with these genes’ susceptibility to alternative splicing [73]. Interestingly, the genes mostly associated with COPD, like SERPINA1 and AGER, showed highly regulated expression of splice variants in lung tissue [74][75], and splice variants were replaced with alternative splicing products. This link of COPD-associated genes may be one possible reason behind the development and progression of the disease.
References
- May, S.M.; Li, J.T.C. Burden of Chronic Obstructive Pulmonary Disease: Healthcare Costs and Beyond. Allergy Asthma Proc. 2015, 36, 4–10.
- Krishnan, A.; Turner, A.M. Chronic Obstructive Pulmonary Disease: The Present and Future. Biomedicines 2022, 10, 499.
- Bhatt, S.P.; Kim, Y.; Harrington, K.F.; Hokanson, J.E.; Lutz, S.M.; Cho, M.H.; DeMeo, D.L.; Wells, J.M.; Make, B.J.; Rennard, S.I.; et al. Smoking Duration Alone Provides Stronger Risk Estimates of Chronic Obstructive Pulmonary Disease than Pack-Years. Thorax 2018, 73, 414–421.
- Diette, G.B.; Accinelli, R.A.; Balmes, J.R.; Buist, A.S.; Checkley, W.; Garbe, P.; Hansel, N.N.; Kapil, V.; Gordon, S.; Lagat, D.K.; et al. Obstructive lung disease and exposure to burning biomass fuel in the indoor environment. Glob. Heart 2012, 7, 265–270.
- Singh, D.; Agusti, A.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Criner, G.J.; Frith, P.; Halpin, D.M.G.; Han, M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: The GOLD Science Committee Report 2019. Eur. Respir. J. 2019, 53, 1900164.
- Lopez, A.D.; Murray, C.C.J.L. The Global Burden of Disease, 1990–2020. Nat. Med. 1998, 4, 1241–1243.
- Qureshi, H.; Sharafkhaneh, A.; Hanania, N.A. Chronic Obstructive Pulmonary Disease Exacerbations: Latest Evidence and Clinical Implications. Ther. Adv. Chronic. Dis. 2014, 5, 212–227.
- Bednarek, M.; Maciejewski, J.; Wozniak, M.; Kuca, P.; Zielinski, J. Prevalence, Severity and Underdiagnosis of COPD in the Primary Care Setting. Thorax 2008, 63, 402–407.
- Denden, S.; Khelil, A.H.; Knani, J.; Lakhdar, R.; Perrin, P.; Lefranc, G.; Chibani, J.B. Alpha-1 Antitrypsin Gene Polymorphism in Chronic Obstructive Pulmonary Disease (COPD). Genet. Mol. Biol. 2010, 33, 23–26.
- Molloy, K.; Hersh, C.P.; Morris, V.B.; Carroll, T.P.; O’Connor, C.A.; Lasky-Su, J.A.; Greene, C.M.; O’Neill, S.J.; Silverman, E.K.; McElvaney, N.G. Clarification of the Risk of Chronic Obstructive Pulmonary Disease in A1-Antitrypsin Deficiency PiMZ Heterozygotes. Am. J. Respir. Crit. Care Med. 2014, 189, 419–427.
- Wells, J.M.; Parker, M.M.; Oster, R.A.; Bowler, R.P.; Dransfield, M.T.; Bhatt, S.P.; Cho, M.H.; Kim, V.; Curtis, J.L.; Martinez, F.J.; et al. Elevated Circulating MMP-9 Is Linked to Increased COPD Exacerbation Risk in SPIROMICS and COPDGene. JCI Insight 2018, 3, e123614.
- Bentley, A.R.; Emrani, P.; Cassano, P.A. Genetic Variation and Gene Expression in Antioxidant Related Enzymes and Risk of COPD: A Systematic Review. Thorax 2008, 63, 956–961.
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632.
- Ganbold, C.; Jamiyansuren, J.; Tumurbaatar, A.; Bayarmaa, A.; Enebish, T.; Dashtseren, I.; Jav, S. The Cumulative Effect of Gene–Gene Interactions Between GSTM1, CHRNA3, CHRNA5 and SOD3 Gene Polymorphisms Combined with Smoking on COPD Risk. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 2857–2868.
- Brøgger, J.; Steen, V.M.; Eiken, H.G.; Gulsvik, A.; Bakke, P. Genetic Association between COPD and Polymorphisms in TNF, ADRB2 and EPHX1. Eur. Respir. J. 2006, 27, 682–688.
- Sparrow, D.; Glynn, R.J.; Cohen, M.; Weiss, S.T. The Relationship of the Peripheral Leukocyte Count and Cigarette Smoking to Pulmonary Function among Adult Men. Chest 1984, 86, 383–386.
- Saha, S.; Brightling, C.E. Eosinophilic Airway Inflammation in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 39–47.
- Cornwell, W.D.; Kim, V.; Song, C.; Rogers, T.J. Pathogenesis of Inflammation and Repair in Advanced COPD. Semin. Respir. Crit. Care Med. 2010, 31, 257–266.
- Polosukhin, V.V.; Gutor, S.S.; Du, R.-H.; Richmond, B.W.; Massion, P.P.; Wu, P.; Cates, J.M.; Sandler, K.L.; Rennard, S.I.; Blackwell, T.S. Small Airway Determinants of Airflow Limitation in Chronic Obstructive Pulmonary Disease. Thorax 2021, 76, 1079–1088.
- Wu, C.-W.; Yau, T.; Fulgar, C.C.; Mack, S.M.; Revilla, A.M.; Kenyon, N.J.; Pinkerton, K.E. Long-Term Sequelae of Smoking and Cessation in Spontaneously Hypertensive Rats. Toxicol. Pathol. 2020, 48, 422–436.
- Janssens, J.-P. Aging of the Respiratory System: Impact on Pulmonary Function Tests and Adaptation to Exertion. Clin. Chest Med. 2005, 26, 469–484.
- Silverman, E.K. Applying Functional Genomics to Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2018, 15, S239–S242.
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 18, 605–607.
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431.
- Ekeowa, U.I.; Gooptu, B.; Belorgey, D.; Hägglöf, P.; Karlsson-Li, S.; Miranda, E.; Pérez, J.; MacLeod, I.; Kroger, H.; Marciniak, S.J.; et al. Alpha1-Antitrypsin Deficiency, Chronic Obstructive Pulmonary Disease and the Serpinopathies. Clin. Sci. (Lond.) 2009, 116, 837–850.
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259.
- Sun, W.; Kechris, K.; Jacobson, S.; Drummond, M.B.; Hawkins, G.A.; Yang, J.; Chen, T.-H.; Quibrera, P.M.; Anderson, W.; Barr, R.G.; et al. Common Genetic Polymorphisms Influence Blood Biomarker Measurements in COPD. PLoS Genet. 2016, 12, e1006011.
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A Genome-Wide Association Study in Chronic Obstructive Pulmonary Disease (COPD): Identification of Two Major Susceptibility Loci. PLoS Genet. 2009, 5, e1000421.
- Cho, M.H.; McDonald, M.-L.N.; Zhou, X.; Mattheisen, M.; Castaldi, P.J.; Hersh, C.P.; Demeo, D.L.; Sylvia, J.S.; Ziniti, J.; Laird, N.M.; et al. Risk Loci for Chronic Obstructive Pulmonary Disease: A Genome-Wide Association Study and Meta-Analysis. Lancet Respir. Med. 2014, 2, 214–225.
- Wilk, J.B.; Shrine, N.R.G.; Loehr, L.R.; Zhao, J.H.; Manichaikul, A.; Lopez, L.M.; Smith, A.V.; Heckbert, S.R.; Smolonska, J.; Tang, W.; et al. Genome-Wide Association Studies Identify CHRNA5/3 and HTR4 in the Development of Airflow Obstruction. Am. J. Respir. Crit. Care Med. 2012, 186, 622–632.
- Minematsu, N.; Nakamura, H.; Iwata, M.; Tateno, H.; Nakajima, T.; Takahashi, S.; Fujishima, S.; Yamaguchi, K. Association of CYP2A6 Deletion Polymorphism with Smoking Habit and Development of Pulmonary Emphysema. Thorax 2003, 58, 623–628.
- Cho, M.H.; Castaldi, P.J.; Wan, E.S.; Siedlinski, M.; Hersh, C.P.; Demeo, D.L.; Himes, B.E.; Sylvia, J.S.; Klanderman, B.J.; Ziniti, J.P.; et al. A Genome-Wide Association Study of COPD Identifies a Susceptibility Locus on Chromosome 19q13. Hum. Mol. Genet. 2012, 21, 947–957.
- Siedlinski, M.; Cho, M.H.; Bakke, P.; Gulsvik, A.; Lomas, D.A.; Anderson, W.; Kong, X.; Rennard, S.I.; Beaty, T.H.; Hokanson, J.E.; et al. Genome-Wide Association Study of Smoking Behaviours in Patients with COPD. Thorax 2011, 66, 894–902.
- Bloom, A.J.; Baker, T.B.; Chen, L.-S.; Breslau, N.; Hatsukami, D.; Bierut, L.J.; Goate, A. Variants in Two Adjacent Genes, EGLN2 and CYP2A6, Influence Smoking Behavior Related to Disease Risk via Different Mechanisms. Hum. Mol. Genet. 2014, 23, 555–561.
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bossé, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; Wyss, A.B.; et al. Genetic Loci Associated with Chronic Obstructive Pulmonary Disease Overlap with Loci for Lung Function and Pulmonary Fibrosis. Nat. Genet. 2017, 49, 426–432.
- Sakornsakolpat, P.; Prokopenko, D.; Lamontagne, M.; Reeve, N.F.; Guyatt, A.L.; Jackson, V.E.; Shrine, N.; Qiao, D.; Bartz, T.M.; Kim, D.K.; et al. Genetic Landscape of Chronic Obstructive Pulmonary Disease Identifies Heterogeneous Cell Type and Phenotype Associations. Nat. Genet. 2019, 51, 494–505.
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial Genes and Pathways in Inflammatory Bowel Disease. Nat. Rev. Microbiol. 2019, 17, 497–511.
- Jin, B.; Li, Y.; Robertson, K.D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617.
- de Vries, M.; van der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; van Duijn, C.M.; Brandsma, C.-A.; van Diemen, C.C.; Vonk, J.M.; et al. From Blood to Lung Tissue: Effect of Cigarette Smoke on DNA Methylation and Lung Function. Respir. Res. 2018, 19, 212.
- Armstrong, D.A.; Chen, Y.; Dessaint, J.A.; Aridgides, D.S.; Channon, J.Y.; Mellinger, D.L.; Christensen, B.C.; Ashare, A. DNA Methylation Changes in Regional Lung Macrophages Are Associated with Metabolic Differences. Immunohorizons 2019, 3, 274–281.
- Peng, H.; Guo, T.; Chen, Z.; Zhang, H.; Cai, S.; Yang, M.; Chen, P.; Guan, C.; Fang, X. Hypermethylation of Mitochondrial Transcription Factor A Induced by Cigarette Smoke Is Associated with Chronic Obstructive Pulmonary Disease. Exp. Lung. Res. 2019, 45, 101–111.
- Rider, C.F.; Carlsten, C. Air Pollution and DNA Methylation: Effects of Exposure in Humans. Clin. Epigenetics 2019, 11, 131.
- Lee, M.K.; Xu, C.-J.; Carnes, M.U.; Nichols, C.E.; Ward, J.M.; BIOS consortium; Kwon, S.O.; Kim, S.-Y.; Kim, W.J.; London, S.J. Genome-Wide DNA Methylation and Long-Term Ambient Air Pollution Exposure in Korean Adults. Clin. Epigenet. 2019, 11, 37.
- Clifford, R.L.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; Fisher, A.J.; Brandsma, C.-A.; Nair, P.; Kobor, M.S.; Hackett, T.-L.; et al. Altered DNA Methylation Is Associated with Aberrant Gene Expression in Parenchymal but Not Airway Fibroblasts Isolated from Individuals with COPD. Clin. Epigenet. 2018, 10, 32.
- Busch, R.; Hobbs, B.D.; Zhou, J.; Castaldi, P.J.; McGeachie, M.J.; Hardin, M.E.; Hawrylkiewicz, I.; Sliwinski, P.; Yim, J.-J.; Kim, W.J.; et al. Genetic Association and Risk Scores in a Chronic Obstructive Pulmonary Disease Meta-Analysis of 16,707 Subjects. Am. J. Respir. Cell Mol. Biol. 2017, 57, 35–46.
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-Antitrypsin Deficiency: Genetic Variations, Clinical Manifestations and Therapeutic Interventions. Mutat. Res. Rev. Mutat. Res. 2017, 773, 14–25.
- Tian, L.; Fong, M.P.; Wang, J.J.; Wei, N.E.; Jiang, H.; Doerge, R.W.; Chen, Z.J. Reversible Histone Acetylation and Deacetylation Mediate Genome-Wide, Promoter-Dependent and Locus-Specific Changes in Gene Expression during Plant Development. Genetics 2005, 169, 337–345.
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Adcock, I.M.; Tsaprouni, L.; Bhavsar, P.; Ito, K. Epigenetic Regulation of Airway Inflammation. Curr. Opin. Immunol. 2007, 19, 694–700.
- Szulakowski, P.; Crowther, A.J.L.; Jiménez, L.A.; Donaldson, K.; Mayer, R.; Leonard, T.B.; MacNee, W.; Drost, E.M. The Effect of Smoking on the Transcriptional Regulation of Lung Inflammation in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 174, 41–50.
- Tao, F.; Zhou, Y.; Wang, M.; Wang, C.; Zhu, W.; Han, Z.; Sun, N.; Wang, D. Metformin Alleviates Chronic Obstructive Pulmonary Disease and Cigarette Smoke Extract-Induced Glucocorticoid Resistance by Activating the Nuclear Factor E2-Related Factor 2/Heme Oxygenase-1 Signaling Pathway. Korean J. Physiol. Pharmacol. 2022, 26, 95–111.
- Ding, J.; Li, F.; Cong, Y.; Miao, J.; Wu, D.; Liu, B.; Wang, L. Trichostatin A Inhibits Skeletal Muscle Atrophy Induced by Cigarette Smoke Exposure in Mice. Life Sci. 2019, 235, 116800.
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette Smoke Induces Distinct Histone Modifications in Lung Cells: Implications for the Pathogenesis of COPD and Lung Cancer. J. Proteome Res. 2014, 13, 982–996.
- Mohammad, N.S.; Nazli, R.; Zafar, H.; Fatima, S. Effects of Lipid Based Multiple Micronutrients Supplement on the Birth Outcome of Underweight Pre-Eclamptic Women: A Randomized Clinical Trial. Pak. J. Med. Sci. 2022, 38, 219–226.
- Nedeljkovic, I.; Carnero-Montoro, E.; Lahousse, L.; van der Plaat, D.A.; de Jong, K.; Vonk, J.M.; van Diemen, C.C.; Faiz, A.; van den Berge, M.; Obeidat, M.; et al. Understanding the Role of the Chromosome 15q25.1 in COPD through Epigenetics and Transcriptomics. Eur. J. Hum. Genet. 2018, 26, 709–722.
- Sohal, S.S.; Walters, E.H. Role of Epithelial Mesenchymal Transition (EMT) in Chronic Obstructive Pulmonary Disease (COPD). Respir. Res. 2013, 14, 120.
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. β-Catenin, Twist and Snail: Transcriptional Regulation of EMT in Smokers and COPD, and Relation to Airflow Obstruction. Sci. Rep. 2017, 7, 10832.
- Aloufi, N.; Alluli, A.; Eidelman, D.H.; Baglole, C.J. Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 11963.
- DeMeo, D.L.; Mariani, T.; Bhattacharya, S.; Srisuma, S.; Lange, C.; Litonjua, A.; Bueno, R.; Pillai, S.G.; Lomas, D.A.; Sparrow, D.; et al. Integration of Genomic and Genetic Approaches Implicates IREB2 as a COPD Susceptibility Gene. Am. J. Hum. Genet. 2009, 85, 493–502.
- Costa, F.F. Non-Coding RNAs: Meet Thy Masters. Bioessays 2010, 32, 599–608.
- Qi, X.; Chen, H.; Fu, B.; Huang, Z.; Mou, Y.; Liu, J.; Xu, Y.; Xiong, W.; Cao, Y. LncRNAs NR-026690 and ENST00000447867 Are Upregulated in CD4+ T Cells in Patients with Acute Exacerbation of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 699–711.
- Donaldson, A.; Natanek, S.A.; Lewis, A.; Man, W.D.-C.; Hopkinson, N.S.; Polkey, M.I.; Kemp, P.R. Increased Skeletal Muscle-Specific MicroRNA in the Blood of Patients with COPD. Thorax 2013, 68, 1140–1149.
- Sun, Y.; An, N.; Li, J.; Xia, J.; Tian, Y.; Zhao, P.; Liu, X.; Huang, H.; Gao, J.; Zhang, X. MiRNA-206 Regulates Human Pulmonary Microvascular Endothelial Cell Apoptosis via Targeting in Chronic Obstructive Pulmonary Disease. J. Cell. Biochem. 2019, 120, 6223–6236.
- Chatila, W.M.; Criner, G.J.; Hancock, W.W.; Akimova, T.; Moldover, B.; Chang, J.-K.; Cornwell, W.; Santerre, M.; Rogers, T.J. Blunted Expression of MiR-199a-5p in Regulatory T Cells of Patients with Chronic Obstructive Pulmonary Disease Compared to Unaffected Smokers. Clin. Exp. Immunol. 2014, 177, 341–352.
- Velasco-Torres, Y.; Ruiz-López, V.; Pérez-Bautista, O.; Buendía-Roldan, I.; Ramírez-Venegas, A.; Pérez-Ramos, J.; Falfán-Valencia, R.; Ramos, C.; Montaño, M. MiR-34a in Serum Is Involved in Mild-to-Moderate COPD in Women Exposed to Biomass Smoke. BMC Pulm. Med. 2019, 19, 227.
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabón, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial Iron Chelation Ameliorates Cigarette Smoke-Induced Bronchitis and Emphysema in Mice. Nat. Med. 2016, 22, 163–174.
- Sun, J.; Gu, X.; Wu, N.; Zhang, P.; Liu, Y.; Jiang, S. Human Antigen R Enhances the Epithelial-Mesenchymal Transition via Regulation of ZEB-1 in the Human Airway Epithelium. Respir. Res. 2018, 19, 109.
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-Transcriptional Regulation of Programmed Cell Death 4 (PDCD4) MRNA by the RNA-Binding Proteins Human Antigen R (HuR) and T-Cell Intracellular Antigen 1 (TIA1). J. Biol. Chem. 2015, 290, 3468–3487.
- Angulo, M.; Lecuona, E.; Sznajder, J.I. Role of MicroRNAs in Lung Disease. Arch. Bronconeumol. 2012, 48, 325–330.
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.M.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56.
- Lackey, L.; McArthur, E.; Laederach, A. Increased Transcript Complexity in Genes Associated with Chronic Obstructive Pulmonary Disease. PLoS ONE 2015, 10, e0140885.
- Zhang, J.; Kuo, C.C.J.; Chen, L. GC Content around Splice Sites Affects Splicing through Pre-MRNA Secondary Structures. BMC Genom. 2011, 12, 90.
- Corley, M.; Solem, A.; Phillips, G.; Lackey, L.; Ziehr, B.; Vincent, H.A.; Mustoe, A.M.; Ramos, S.B.V.; Weeks, K.M.; Moorman, N.J.; et al. An RNA Structure-Mediated, Posttranscriptional Model of Human α-1-Antitrypsin Expression. Proc. Natl. Acad. Sci. USA 2017, 114, E10244–E10253.
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, Classification, and Expression of RAGE Gene Splice Variants. FASEB J. 2008, 22, 1572–1580.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
814
Revisions:
2 times
(View History)
Update Date:
21 Jun 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No