Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mariana Pinto | -- | 2046 | 2023-06-20 14:40:45 | | | |
| 2 | Conner Chen | Meta information modification | 2046 | 2023-06-25 04:31:08 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pinto, M.T.; Eiras Martins, G.; Vieira, A.G.S.; Galvão, J.M.S.; De Pádua Souza, C.; Macedo, C.R.P.D.; Lopes, L.F. Etiopathogenesis of Ovarian Germ Cell Tumors. Encyclopedia. Available online: https://encyclopedia.pub/entry/45866 (accessed on 24 June 2026).
Pinto MT, Eiras Martins G, Vieira AGS, Galvão JMS, De Pádua Souza C, Macedo CRPD, et al. Etiopathogenesis of Ovarian Germ Cell Tumors. Encyclopedia. Available at: https://encyclopedia.pub/entry/45866. Accessed June 24, 2026.
Pinto, Mariana Tomazini, Gisele Eiras Martins, Ana Glenda Santarosa Vieira, Janaina Mello Soares Galvão, Cristiano De Pádua Souza, Carla Renata Pacheco Donato Macedo, Luiz Fernando Lopes. "Etiopathogenesis of Ovarian Germ Cell Tumors" Encyclopedia, https://encyclopedia.pub/entry/45866 (accessed June 24, 2026).
Pinto, M.T., Eiras Martins, G., Vieira, A.G.S., Galvão, J.M.S., De Pádua Souza, C., Macedo, C.R.P.D., & Lopes, L.F. (2023, June 20). Etiopathogenesis of Ovarian Germ Cell Tumors. In Encyclopedia. https://encyclopedia.pub/entry/45866
Pinto, Mariana Tomazini, et al. "Etiopathogenesis of Ovarian Germ Cell Tumors." Encyclopedia. Web. 20 June, 2023.
Copy Citation
Neoplasms arising in the ovary originate from different cell types which constitute the tissue of the ovary. The surface epithelium, the stroma, and the cellular elements of the follicle may give rise to distinct tumors; in particular, the cellular elements of the follicle can result in sex cord-stromal tumors or germ cell tumors. OGCTs occur due to the pathologic transformation of the primordial germ cell (PGC) during the distinct stages of embryonic development.
ovarian cancer
germ cell tumors
1. Introduction
Germ cell tumors (GCTs) comprise a heterogeneous group of benign and malignant neoplasms. They are either located in the gonads (ovary or testis) or they are extragonadal. Clinically, histopathologically, genetically, and molecularly, these tumors vary significantly [1].
Germ cell & gonadal tumors are rare in adults; indeed, they occur predominantly in children, adolescents, and young adults. Moreover, they account for approximately 10% of cancer diagnoses in 15–19 years old group [2]. Ovarian germ cell tumors (OGCTs) are usually detected at an early stage, and they are associated with a favorable prognosis; however, the advanced stages of the disease, and cases where the patient relapses, show the long-term toxicity of common platinum-based regimens [3][4].
In recent decades, major efforts have been made to find new therapeutic alternatives to the platinum-based treatments, to understand the biology of GCTs, and to understand the molecular mechanisms of GCTs. Several studies have been performed in order to provide novel insights into integrated genomic analysis, microRNAs, DNA methylation, the molecular implications of treatment resistance, and the development of in vitro and in vivo models. The malignant germ cells that are found in the ovary often present numerous chromosomal variations and genetic abnormalities that are unrelated to the clinical findings. The molecular characterization of patients’ tumors may lead to the identification of prognostic and biomarkers.
2. Etiopathogenesis of Ovarian Germ Cell Tumors
Neoplasms arising in the ovary originate from different cell types which constitute the tissue of the ovary. The surface epithelium, the stroma, and the cellular elements of the follicle may give rise to distinct tumors; in particular, the cellular elements of the follicle can result in sex cord-stromal tumors or germ cell tumors [5]. OGCTs occur due to the pathologic transformation of the primordial germ cell (PGC) during the distinct stages of embryonic development (Figure 1).
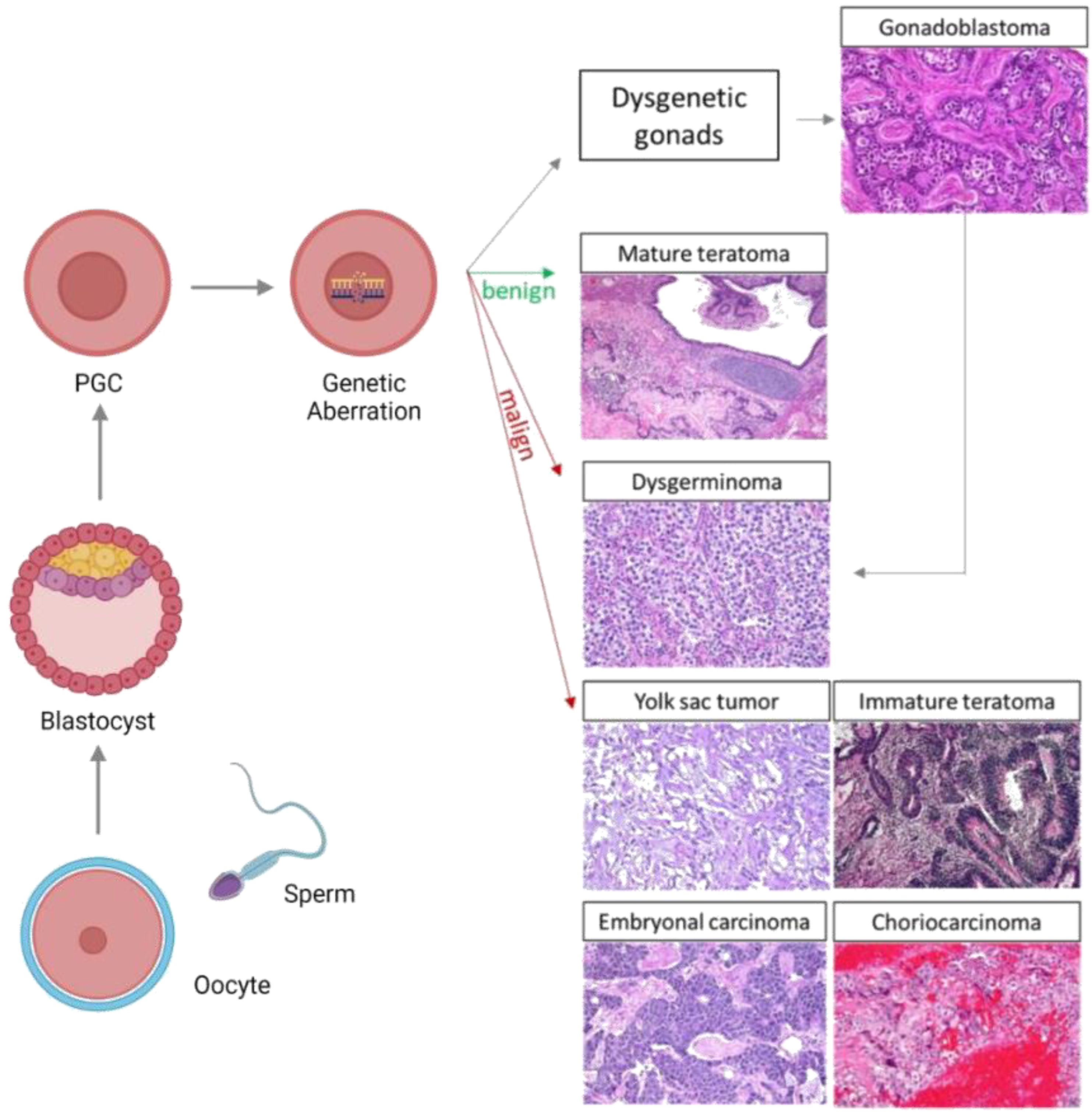
Figure 1. Patients with disordered female gonadal development, caused by various genomic aberrations, can present gonadal dysgenesis with a high risk of developing malignant OGCTs; such tumors often develop from gonadoblastomas. Genomic abnormalities in PGCs can also develop into benign tumors as mature teratomas, and malign tumors including dysgerminomas, yolk sac tumors, immature teratomas, embryonal carcinoma, and choriocarcinoma may also develop. The green arrows represent benign tumors and the red arrows represent malign tumors. PGC: Primordial germ cell.
The etiopathogenesis of OGCTs remains poorly understood; the cellular and molecular mechanisms of testicular germ cell tumors (TGCTs) have been studied to a greater extent. Both types of germ cell tumors are markedly aneuploid [6][7], and DNA methylation aberrations, the number of copied abnormalities, and mutations can contribute to the emergence of GCTs [8].
GCTs are a diverse group which is presumed to have a common cell of origin—the primordial germ cell (PGC). These tumors emerge in various midline or near-midline sites, and they are thought to be due to the arrested or aberrant migration of PGCs during embryogenesis. Moreover, the incidence and distribution of GCTs proves the importance of regulated cell death in midline sites [9].
2.1. Gonadal Dysgenesis
Disorders of sex development (DSD) comprise a series of diverse pathologies that are characterized by congenital conditions in which the development of chromosomal, gonadal, or anatomical sex is abnormal, thus resulting in the malformation of the internal and/or external genital organs [10][11]. DSD patients with gonadal dysgenesis (GD) present different somatic and genetic features with the development of hypoplastic genitalia [12][13]. Patients with GD and genotype 46, XY are at an increased risk of developing malignant OGCTs, which often emerge from gonadoblastomas (GB—a rare tumor containing both germ cells and sex cord-stromal cells) [4][13][14][15]. In such cases, the occurrence of Y chromosomal material confers a 30–40% risk of GB, and moreover, 50% of gonadoblastomas are correlated with dysgerminomas [16][17].
The emergence of gonadoblastomas appears to be related to the TSPY gene, which is located on the short arm of the Y chromosome; it is responsible for encoding testis-specific protein-Y (TSPY) [18][19]. High levels of TSPY support the survival and proliferation of immature PGC, which remains in an embryonic state, with an increased expression of OCT3/4 [13][20].
Germ cells from patients with GD might escape cell death if there is a persistent expression of both OCT3/4 and TSPY; this is because it may later give rise to clonal expansion and neoplastic formation. Hermus et al. reported that 17 out of 19 (89%) of gonadoblastoma cases (with or without present dysgerminoma) showed positive staining with regard to the TSPY protein in neoplastic cells [18].
2.2. General Features of Embryonic and Gonadal Development
An understanding of gonad development and the characteristics of germ cells at different stages of differentiation is fundamental for comprehending the origin and features of several types of GCTs, particularly OGCTs [21].
The integration of three main events leads to PGC specification. These events are as follows: repression of the somatic program, reacquisition of potential pluripotency, and genome-wide epigenetic reprogramming [5][22]. In the embryonic phase, PGCs develop in the yolk sacs of embryos, and they migrate to the gonads through the midline of the body in a process controlled by the stem cell growth factor receptor, KIT (KIT), and its ligand, KIT ligand (KITLG) [5][8]. Embryonic stem cells (ESCs), PGCs, and germ cells are proliferating cells containing active telomerase, and they are able to replicate indefinitely. In addition to potentially infinite replicative abilities, PGCs share other hallmark characteristics with cancer cells, such as anaerobic glycolysis and the ability to migrate [22][23]. A feature of such ESCs is the expression of OCT4, SOX2, and NANOG, which maintain pluripotency [24].
Runyan et al. showed that the process of targeted migration and controlled cell death are essential for the possible localization of germ cells in the genital ridges. The study showed that the regulation of apoptosis during migration causes the removal of midline germ cells and those pro-apoptotic genes of the intrinsic pathway, which are upregulated in migratory germ cells [25].
ESCs that are derived from the preimplantation inner cell mass (ICM) and epiblast, which present totipotent developmental potential (so-called naïve state), are distinguished by a permissive epigenetic signature, as well as two active X chromosomes (in female cells) [22][24]. The expression of OCT4 is driven by a distal enhancer, and OCT4 partners with SOX2 (SRY-box 2). Between human embryonic weeks three and four, these cells enter a primed state by submitting female X inactivation and promoting methylation in pluripotency genes, thus implementing a restricted self-renewal ability and pluripotent developmental potential. During the same period, PGCs are specified, and are subsequently the only OCT4-expressing cells in the embryo. During human PGC migration, approximately weeks five and six, OCT4 switches SOX2 for SOX17 (SRY-box 17). Moreover, SOX17 and BLIMP1 (B lymphocyte-induced maturation protein 1) are also important for maintaining the wellbeing of PGCs by preventing them from being reprogrammed to an ESC [22][24][26].
When PGCs are in a latent potency state, which is determined by their epigenetic status, they can be reprogrammed to enter a primed state. During GCT development, ESCs may enter a naïve state. In addition, all the GCT subtypes present the same global methylation and genomic imprinting patterns of the GCT stem cells, which strongly resemble those of their normal counterparts [22]. Imprinted genes are localized within various chromosomal clusters that contain GC-rich regions with differentially methylated CpG dinucleotides. Genomic imprinting patterns are responsible for allele-specific gene expression. Imprinted genes in mammals have specific roles to play in the developing germ cells. As opposed to to somatic cells, which maintain the parent-specific imprinting pattern, germ cells must, at some stage, erase the imprinted genes and establish a new, sex-specific imprinting template [27][28].
Once PGCs reach the bipotential gonads, the absence of the SRY gene, which is typically located on the Y chromosome or its downstream target SOX9, and the subsequent activity of female-specific genes, promote the development of the gonads into ovaries. A subset of ovarian-specific genes controls the ovarian morphogenesis process [21]. In fetal ovaries, at around the tenth week of gestation, the retinoic acid produced by the mesonephros stimulates the initiation of meiosis, which appears to occur asynchronously. Indeed, as increasing numbers of germ cells initiate meiosis, some oogonia still express stem cell markers and continue to proliferate until at least week 16 [5][21]. After the 12th week, the oogonia seem to segment into two cell populations: a KIT/OCT4 subset located at the periphery and a VASA positive subset that is located near the center of the ovary. A few genes control the mitotic-to-meiotic transition, and any dysfunction during this transitional period can be a root cause of the conversion of PGCs into GCTs [21].
Following gonadal colonization, proliferating oogonia arrange themselves into cyst-like structures with supporting pregranulosa cells by the end of the seventh month of gestation. Furthermore, at this point, they rapidly lose their POU domain, as well as their class 5 and transcription factor 1 (POU5F1) expression, and mitotic activity is terminated. At this stage, almost all oogonia enter meiotic prophase I and become primary oocytes, where they remain arrested, and they subsequently form primordial follicles [5][21]. Ovary differentiation occurs due to feminizing factors WNT4 and FOXL2; the latter factor has been termed the gatekeeper of ovarian identity [5][22].
2.3. OGCT Development
GCTs are rarely caused by somatic driver mutations; indeed, they are the result of reprogramming PGCs due to a failure in the cell process to control their latent developmental potency. This not only explains their developmental potential, but it also describes the diverse clinical and pathological aspects of GCTs [22].
In accordance with the model of tumorigenesis proposed by Teilum, germinomas (dysgerminomas in ovarian sites) emerge directly from primordial germ cells, and consequently, they retain their pluripotent state. Embryonal carcinomas (ECs) presenting early embryonic differentiation can give rise to tumors containing all three germ layers (endoderm, ectoderm, and mesoderm). In contrast, PGCs that exhibit extra-embryonic differentiation result in either yolk-sac tumors (YSTs) or choriocarcinomas (CC). The mixed germ cell tumors contain various malignant histologies [29][30]. Dysgerminoma and immature teratoma are the most common types of OGCT, comprising 65–70% of all OGCTs, followed by YSTs (14.5%), and finally, mixed GCTs (5.3%) [31][32].
Most OGCTs are unilateral; however, 10–15% of dysgerminoma and 5–10% of the mixed OGCT subtype may be bilateral [33][34].
Furthermore, PGCs expressing pluripotent genes, such as NANOG and POU5F1, and which express complete demethylation, gain additional genomic abnormalities, such as the KIT mutation, or the isochromosome 12p; subsequently, this causes the emergence of dysgerminomas (DGs). PGCs expressing DPPA3 sometimes gain isochromosome 12p and they subsequently develop into ECs. PGCs expressing DPPA3 restore DNA methylation and differentiate into sperm or egg cells. During differentiation, further genomic abnormalities are acquired by PGCs, which can develop into YSTs or teratomas. Teratomas, ECs, and YSTs imprint in a sex-specific manner [8].
Pediatric and adult GCTs differ from the imprinting patterns of loci in, for example, IGF2/H19; this means that pediatric GCTs tend to emerge from more immature PGCs. In addition, compared with germinomas, pediatric YSTs show increased methylation in several genes’ regulatory loci, and they demonstrate a methylator phenotype, such as a decrease in the number of genes that programme cell death and repress WNT signaling [27][30].
Kato et al. investigated genetic zygosity in a series of mature ovarian teratomas, struma ovarii, and ovarian carcinoids. They showed that homozygous genotypes were present in 50% of mature teratomas, 50% of struma ovarii, and 33% of ovarian carcinoids; this suggests that the oocyte that emerges after meiosis I is an important factor that causes these tumors [6].
Multi-region whole exome sequencing of immature ovarian teratomas (in the range of 8–29 years) was performed, and the results revealed that this type of tumor is characterized by 2N near-diploid genomes which have undergone a severe loss in heterozygosity. In addition, they exhibit an absence of the genes which harbor recurrent mutations or known oncogenic variants. Moreover, different patterns in the left and right ovaries displayed a loss in heterozygosity, thus suggesting that bilateral ovarian teratomas emerge independently of one another. Altogether, these outcomes show that numerous meiotic mistakes can form genetically distinct tumors; these tumors are unique as a result of their stark allelic imbalances, a lack of somatic mutations, and copy number alterations [35].
References
- Sessa, C.; Schneider, D.T.; Planchamp, F.; Baust, K.; Braicu, E.I.; Concin, N.; Godzinski, J.; McCluggage, W.G.; Orbach, D.; Pautier, P.; et al. ESGO–SIOPE Guidelines for the Management of Adolescents and Young Adults with Non-Epithelial Ovarian Cancers. Lancet Oncol. 2020, 21, e360–e368.
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. Non-Epithelial Ovarian Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv1–iv18.
- Maoz, A.; Matsuo, K.; Ciccone, M.A.; Matsuzaki, S.; Klar, M.; Roman, L.D.; Sood, A.K.; Gershenson, D.M. Molecular Pathways and Targeted Therapies for Malignant Ovarian Germ Cell Tumors and Sex Cord–Stromal Tumors: A Contemporary Review. Cancers 2020, 12, 1398.
- Kraggerud, S.M.; Hoei-Hansen, C.E.; Alagaratnam, S.; Skotheim, R.I.; Abeler, V.M.; Rajpert-De Meyts, E.; Lothe, R.A. Molecular Characteristics of Malignant Ovarian Germ Cell Tumors and Comparison with Testicular Counterparts: Implications for Pathogenesis. Endocr. Rev. 2013, 34, 339–376.
- Kato, N.; Sakamoto, K.; Murakami, K.; Iwasaki, Y.; Kamataki, A.; Kurose, A. Genetic Zygosity of Mature Ovarian Teratomas, Struma Ovarii, and Ovarian Carcinoids. Virchows Arch. 2018, 473, 177–182.
- Van Nieuwenhuysen, E.; Busschaert, P.; Neven, P.; Han, S.N.; Moerman, P.; Liontos, M.; Papaspirou, M.; Kupryjanczyk, J.; Hogdall, C.; Hogdall, E.; et al. The Genetic Landscape of 87 Ovarian Germ Cell Tumors. Gynecol. Oncol. 2018, 151, 61–68.
- Kubota, Y.; Seki, M.; Kawai, T.; Isobe, T.; Yoshida, M.; Sekiguchi, M.; Kimura, S.; Watanabe, K.; Sato-Otsubo, A.; Yoshida, K.; et al. Comprehensive Genetic Analysis of Pediatric Germ Cell Tumors Identifies Potential Drug Targets. Commun. Biol. 2020, 3, 544.
- Palmer, R.D.; Foster, N.A.; Vowler, S.L.; Roberts, I.; Thornton, C.M.; Hale, J.P.; Schneider, D.T.; Nicholson, J.C.; Coleman, N. Malignant Germ Cell Tumours of Childhood: New Associations of Genomic Imbalance. Br. J. Cancer 2007, 96, 667–676.
- Faure-Conter, C.; Orbach, D.; Fresneau, B.; Verité, C.; Bonneau, J.; Thebaud, E.; Poirée, M.; Thouvenin, S.; Pluchart, C.; Mure, P.Y.; et al. Disorder of Sex Development with Germ Cell Tumors: Which Is Uncovered First? Pediatr. Blood Cancer 2020, 67.
- Hughes, I.A. Consensus Statement on Management of Intersex Disorders. Arch. Dis. Child. 2005, 91, 554–563.
- Piazza, M.J.; Urbanetz, A.A. Germ Cell Tumors in Dysgenetic Gonads. Clinics 2019, 74, e408.
- Hennes, E.; Zahn, S.; Lopes, L.; Schönberger, S.; Leuschner, I.; Göbel, U.; Calaminus, G.; Schneider, D. Molecular Genetic Analysis of Bilateral Ovarian Germ Cell Tumors. Klin. Padiatr. 2012, 224, 359–365.
- Duhil de Bénazé, G.; Pacquement, H.; Faure-Conter, C.; Patte, C.; Orbach, D.; Corradini, N.; Berger, C.; Sudour-Bonnange, H.; Vérité, C.; Martelli, H.; et al. Paediatric Dysgerminoma: Results of Three Consecutive French Germ Cell Tumours Clinical Studies (TGM-85/90/95) with Late Effects Study. Eur. J. Cancer 2018, 91, 30–37.
- Capito, C.; Leclair, M.-D.; Arnaud, A.; David, A.; Baron, S.; Corradini, N.; Héloury, Y. 46,XY Pure Gonadal Dysgenesis: Clinical Presentations and Management of the Tumor Risk. J. Pediatr. Urol. 2011, 7, 72–75.
- Lin, K.Y.; Bryant, S.; Miller, D.S.; Kehoe, S.M.; Richardson, D.L.; Lea, J.S. Malignant Ovarian Germ Cell Tumor—Role of Surgical Staging and Gonadal Dysgenesis. Gynecol. Oncol. 2014, 134, 84–89.
- Scully, R.E. Gonadoblastoma. A Review of 74 Cases. Cancer 1970, 25, 1340–1356.
- Hersmus, R.; Stoop, H.; van de Geijn, G.J.; Eini, R.; Biermann, K.; Oosterhuis, J.W.; DHooge, C.; Schneider, D.T.; Meijssen, I.C.; Dinjens, W.N.M.; et al. Prevalence of C-KIT Mutations in Gonadoblastoma and Dysgerminomas of Patients with Disorders of Sex Development (DSD) and Ovarian Dysgerminomas. PLoS ONE 2012, 7, e43952.
- Cools, M.; Stoop, H.; Kersemaekers, A.-M.F.; Drop, S.L.S.; Wolffenbuttel, K.P.; Bourguignon, J.-P.; Slowikowska-Hilczer, J.; Kula, K.; Faradz, S.M.H.; Oosterhuis, J.W.; et al. Gonadoblastoma Arising in Undifferentiated Gonadal Tissue within Dysgenetic Gonads. J. Clin. Endocrinol. Metab. 2006, 91, 2404–2413.
- Looijenga, L.H.J.; Stoop, H.; de Leeuw, H.P.J.C.; de Gouveia Brazao, C.A.; Gillis, A.J.M.; van Roozendaal, K.E.P.; van Zoelen, E.J.J.; Weber, R.F.A.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) Identifies Cells with Pluripotent Potential in Human Germ Cell Tumors. Cancer Res. 2003, 63, 2244–2250.
- Dolci, S.; Campolo, F.; De Felici, M. Gonadal Development and Germ Cell Tumors in Mouse and Humans. Semin. Cell. Dev. Biol. 2015, 45, 114–123.
- Oosterhuis, J.W.; Looijenga, L.H.J. Human Germ Cell Tumours from a Developmental Perspective. Nat. Rev. Cancer 2019, 19, 522–537.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Hackett, J.A.; Surani, M.A. Regulatory Principles of Pluripotency: From the Ground State Up. Cell Stem Cell 2014, 15, 416–430.
- Runyan, C.; Schaible, K.; Molyneaux, K.; Wang, Z.; Levin, L.; Wylie, C. Steel Factor Controls Midline Cell Death of Primordial Germ Cells and Is Essential for Their Normal Proliferation and Migration. Development 2006, 133, 4861–4869.
- Anderson, R.A.; Fulton, N.; Cowan, G.; Coutts, S.; Saunders, P.T. Conserved and Divergent Patterns of Expression of DAZL, VASA and OCT4 in the Germ Cells of the Human Fetal Ovary and Testis. BMC Dev. Biol 2007, 7, 136.
- Schneider, D.T.; Schuster, A.E.; Fritsch, M.K.; Hu, J.; Olson, T.; Lauer, S.; Göbel, U.; Perlman, E.J. Multipoint Imprinting Analysis Indicates a Common Precursor Cell for Gonadal and Nongonadal Pediatric Germ Cell Tumors. Cancer Res. 2001, 61, 7268–7276.
- Ross, J.A.; Schmidt, P.T.; Perentesis, J.P.; Davies, S.M. Genomic Imprinting of H19 and Insulin-like Growth Factor-2 in Pediatric Germ Cell Tumors. Cancer 1999, 85, 1389–1394.
- Teilum, G. Classification of Endodermal Sinus Tumour (Mesoblastoma Vitellinum) and so-called “Embryonal Carcinoma” of the Ovary. Acta Pathol. Microbiol. Scand. 1965, 64, 407–429.
- Shaikh, F.; Murray, M.J.; Amatruda, J.F.; Coleman, N.; Nicholson, J.C.; Hale, J.P.; Pashankar, F.; Stoneham, S.J.; Poynter, J.N.; Olson, T.A.; et al. Paediatric Extracranial Germ-Cell Tumours. Lancet Oncol. 2016, 17, e149–e162.
- Bryant, C.S.; Kumar, S.; Shah, J.P.; Mahdi, H.; Ali-Fehmi, R.; Munkarah, A.R.; Deppe, G.; Morris, R.T. Racial Disparities in Survival among Patients with Germ Cell Tumors of the Ovary—United States. Gynecol. Oncol. 2009, 114, 437–441.
- Smith, H.O.; Berwick, M.; Verschraegen, C.F.; Wiggins, C.; Lansing, L.; Muller, C.Y.; Qualls, C.R. Incidence and Survival Rates for Female Malignant Germ Cell Tumors. Obstet. Gynecol. 2006, 107, 1075–1085.
- Euscher, E.D. Germ Cell Tumors of the Female Genital Tract. Surg. Pathol. Clin. 2019, 12, 621–649.
- Veneris, J.T.; Mahajan, P.; Frazier, A.L. Contemporary Management of Ovarian Germ Cell Tumors and Remaining Controversies. Gynecol. Oncol. 2020, 158, 467–475.
- Heskett, M.B.; Sanborn, J.Z.; Boniface, C.; Goode, B.; Chapman, J.; Garg, K.; Rabban, J.T.; Zaloudek, C.; Benz, S.C.; Spellman, P.T.; et al. Multiregion Exome Sequencing of Ovarian Immature Teratomas Reveals 2N Near-Diploid Genomes, Paucity of Somatic Mutations, and Extensive Allelic Imbalances Shared across Mature, Immature, and Disseminated Components. Mod. Pathol. 2020, 33, 1193–1206.
More
Information
Subjects:
Developmental Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
758
Revisions:
2 times
(View History)
Update Date:
25 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No