Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natticha Sumneang | -- | 1927 | 2023-06-20 05:05:22 | | | |
| 2 | Conner Chen | -25 word(s) | 1902 | 2023-06-25 03:32:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sumneang, N.; Tanajak, P.; Oo, T.T. Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity. Encyclopedia. Available online: https://encyclopedia.pub/entry/45825 (accessed on 09 August 2026).
Sumneang N, Tanajak P, Oo TT. Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity. Encyclopedia. Available at: https://encyclopedia.pub/entry/45825. Accessed August 09, 2026.
Sumneang, Natticha, Pongpan Tanajak, Thura Tun Oo. "Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity" Encyclopedia, https://encyclopedia.pub/entry/45825 (accessed August 09, 2026).
Sumneang, N., Tanajak, P., & Oo, T.T. (2023, June 20). Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity. In Encyclopedia. https://encyclopedia.pub/entry/45825
Sumneang, Natticha, et al. "Toll-like Receptor 4 Inflammatory Perspective on Doxorubicin-Induced Cardiotoxicity." Encyclopedia. Web. 20 June, 2023.
Copy Citation
Doxorubicin (Dox) is one of the most frequently used chemotherapeutic drugs in a variety of cancers, but Dox-induced cardiotoxicity diminishes its therapeutic efficacy. The underlying mechanisms of Dox-induced cardiotoxicity are still not fully understood. More significantly, there are no established therapeutic guidelines for Dox-induced cardiotoxicity. To date, Dox-induced cardiac inflammation is widely considered as one of the underlying mechanisms involved in Dox-induced cardiotoxicity. The Toll-like receptor 4 (TLR4) signaling pathway plays a key role in Dox-induced cardiac inflammation, and growing evidence reports that TLR4-induced cardiac inflammation is strongly linked to Dox-induced cardiotoxicity.
doxorubicin
Toll-like receptor 4
cardiotoxicity
1. Introduction
Cancer is a major public health problem worldwide. Cancer is a complicated disease caused by internal factors (e.g., inherited mutations, hormones, and immune conditions) and environmental factors (e.g., diet, radiation, and infectious organisms) [1][2]. According to a report from the World Health Organization, cancer is one of the leading causes of death worldwide [3]. In the United States of America, there were approximately 1.98 million new cases of diagnosed cancer in 2022 [4]. Furthermore, the number of cancer cases around the world is expected to reach around 26 million by 2030, with 17 million deaths per year [5].
Currently, doxorubicin (Dox) is one of the most used chemotherapeutic drugs for various types of cancer, such as breast, lung, ovarian, and thyroid cancers [6][7]. Although it is able to combat tumor cells, it is also harmful to normal cells, including cardiomyocytes [7][8][9]. In addition, several studies have reported the potential mechanisms of Dox-induced cardiotoxicity mediated by mitochondrial dysfunction, DNA damage, oxidative stress, and apoptosis [2][7][9]. A growing body of research suggests that aseptic inflammation might also play a plausible role in Dox-induced cardiotoxicity due to the activation of the innate immune system after Dox treatment [10][11][12]. A number of studies showed that Dox treatment upregulates the expressions of pro-inflammatory cytokines in cardiac tissue, such as tumor nuclear factor (TNF)-α, interleukin (IL)-1β, and IL-6, via activation of the nuclear factor-κB (NF-κB), triggering the progression of cardiovascular diseases and other adverse cardiac events [13][14][15]. Dox-induced cardiotoxicity mainly limits the cumulative dose of Dox in clinical settings. Cardiotoxicity is a potentially lethal condition and also a well-known adverse effect of Dox; however, the underlying molecular mechanism of Dox-induced cardiotoxicity, particularly related to inflammation, is not fully understood. Although the underlying mechanisms of Dox-induced cardiotoxicity remain complex, the role of cardiac inflammation in Dox-induced cardiotoxicity has become a focus for researchers in recent years.
Toll-like receptor 4 (TLR4), an important member of the TLR family, is part of the innate immune system that responds to the endogenous and exogenous signals and triggers pathophysiological functions in organs, including the heart [16][17][18][19]. The precise molecular mechanisms of TLR4 signaling have been elucidated. Upon binding to a specific ligand with the help of myeloid differentiation factor 2 (MD2), the TLR4 signaling pathway is activated, followed by recruiting the intracellular adaptor molecules and releasing the inflammatory mediators [15][20][21]. Interestingly, there is increasing evidence that Dox-induced TLR4 signaling pathway activation is implicated in cardiac adverse effects, which manifest as left ventricle (LV) impairment [20][22][23]. In support of this evidence, a previous study demonstrated that Dox-induced cardiac adverse effects were completely alleviated in TLR4 knockout mice [22]. Therefore, TLR4 signaling is thought to be involved in the mechanism contributing to Dox-induced cardiotoxicity, and inhibition of TLR4 is considered to be one the potential interventions against Dox-induced cardiomyopathy [22][23].
2. The Mechanism of TLR4 on Dox-Induced Cardiotoxicity
Growing evidence has shown that Dox activates the innate immune system, which is one of the expected components of the response against tumor cells [2][9]. However, Dox-induced innate immune activation provokes the release of inflammatory cytokines in a number of tissues, including the heart, intestine, brain, and liver, which results in inflammation in non-targeted organs [9][10][24][25]. To date, several studies have demonstrated that Dox-induced cardiac inflammation is strongly linked to Dox-induced cardiotoxicity [10][14][26][27]. Dox induces NF-κB activity to promote cardiac inflammation by upregulating TNF-α, IL-1β, and IL-6 expression [15][26].
TLR4 is responsible for the innate immune response; thus, the activation of TLR4 has become one of the most attractive targets in recent years [28][29]. Moreover, the expression of TLR4 is also found in the cardiomyocytes in addition to the immune cells [21]. This receptor responds to exogenous and endogenous ligands: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [28]. TLR4 recognizes the bacterial LPS as a major PAMP with the help of its co-receptor MD2 [21]. In addition, the TLR4 signaling pathway can also be activated by various endogenous DAMPs, namely alarmin protein, including high-mobility group protein box 1 (HMGB1) and the heat shock protein family (Hsps) [28][30]. Upon ligand binding, TLR4 dimerization occurs, followed by activation of the myeloid differentiation primary response protein 88 (MyD88); the downstream signaling pathways propagate NF-κB phosphorylation via reducing the inhibitory κB kinase (IKK) response, leading to upregulation of inflammatory cytokines and cardiac inflammation [21]. In addition to NF-κB activity, components of mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, are also activated as downstream signal transducers of MyD88 to contribute to the regulation of pro-inflammatory responses [18][21].
To date, a growing body of evidence demonstrates that Dox induces the release of PAMP and DAMP, resulting in TLR4-mediated cardiac inflammation that contributes significantly to cardiotoxicity [10][15][28][31]. A simplified overview of the mechanistic activation of TLR4 in Dox-induced cardiotoxicity is shown in Figure 1. Therefore, inhibition of TLR4 would have a therapeutic benefit against Dox-induced cardiotoxicity.
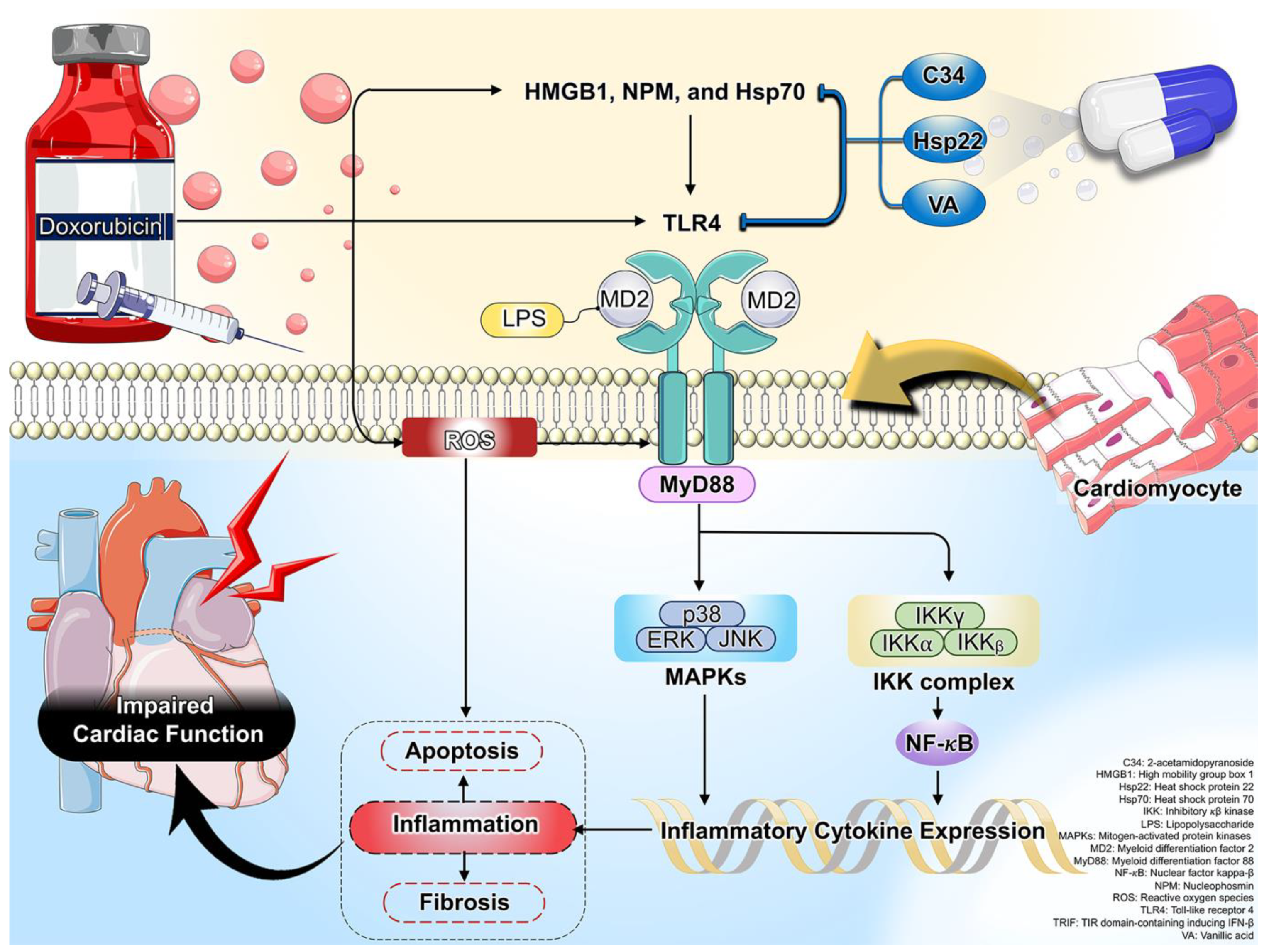
Figure 1. A schematic presentation of oxidative stress and inflammation-related mechanisms via TLR4 in Dox-induced cardiotoxicity. Activation of TLR4 was exhibited by HMGB1, NPM, and Hsp70, leading to increased cardiac inflammation, apoptosis, and fibrosis, and impaired cardiac function in Dox treatment. Its subsequent detrimental effects were effectively attenuated by treatment with C34, Hsp22, and VA.
3. The Effects of Dox on TLR4 Expression in Cardiomyocytes: Reports from In Vitro Studies
To date, the relationship between the role of TLR4 and Dox administration is still scarce; however, increasing evidence from in vitro studies suggests that Dox-mediated cardiotoxicity is related to the upregulation of cardiomyocyte TLR4 expressions [32][33][34]. Li et al. reported that Dox administration upregulated the expression of TLR4 in human cardiomyocyte cell lines [32]. In addition, Feng et al. demonstrated that Dox treatment significantly enhanced the TLR4 signaling pathway in H9c2 cardiomyocytes, as evidenced by increased TLR4 expression and its downstream signaling pathways, including MyD88, NF-κB, IL-1 β, IL-6, and TNF-α, along with decreased IkBα expression [27]. According to these two in vitro results. However, further research is necessary to fully understand the underlying molecular signaling of Dox-induced TLR4 signaling pathway activation in cardiomyocytes.
Traditionally, TLR4 ligands, PAMPs, and DAMPs bind to MD2 to activate the TLR4 signaling pathway in cardiomyocytes, resulting in cardiac inflammation [18][35]. The DNA-binding nuclear protein HMGB1, which regulates gene transcription and nucleosome stability, has a variety of biological functions in clinicopathological conditions [36]. It is considered a cytokine involved in the activation of innate immunity after being actively released from cells or passively released upon cell injury [36]. Interestingly, TLR4 functions as the major HMGB1 receptor [37]. Disulfide HMGB1 activates the TLR4 complex, binding to MD2, which triggers dimerization of TLR4 that can stimulate downstream signal transduction molecules (e.g., NF-κB) to produce pro-inflammatory cytokines [37]. A previous study demonstrated that the release of HMGB1 from the cells was increased following Dox treatment [38]. Moreover, the oxidative stress and DAMPs were considered to be major stimulators of HMGB1 release, activating inflammation through the TLR4 signaling pathway [38]. Therefore, Dox is closely related to altered HMGB1 levels, leading to induced cardiotoxicity.
In addition to HMGB1, there is also nucleophosmin (NPM), which behaves similarly to an alarmin protein released in response to cellular excess due to severe injury [39]. Similar to HMGB1, NPM can also bind to the TLR4 signaling cascade, leading it to exerting pro-inflammatory cytokines [39]. Interestingly, a previous study showed that Dox induced nucleolar stress, subsequently disrupting and releasing the NPM from the nucleolus. Then, the extracellular NPM induced inflammation via TLR4 signaling pathway activation [39]. To date, there is only one study that has shown that NPM can bind to the TLR4 signaling cascade, promoting pro-inflammatory function following the Dox treatment of human cardiac mesenchymal progenitor cells (hCmPCs) [39]. Thus, NPM is a novel ligand of TLR4 that activates inflammation in Dox-treated cardiotoxicity [39].
Inhibition of TLR4 via genetic deletion suppressed Dox-induced cardiotoxicity [27][31][32]. This evidence was provided by previous studies, showing that genetic ablation of TLR4 not only reduced inflammation but also decreased apoptosis in cardiomyocytes after exposure to Dox [27][31][32]. Although molecular signaling through TLR4 has not been demonstrated in the genetic deletion of TLR4 on apoptosis in cardiomyocytes after Dox exposure, the aforementioned discussions suggest that pro-inflammatory cytokines [27], mediated by TLR4, were suppressed, resulting in a decrease in cardiomyocyte apoptosis, as evidenced by the decrease in apoptotic proteins (Bax and cleaved caspase-3) and the increase in anti-apoptotic proteins (Bcl-2) [31][32]. These in vitro studies demonstrate the plausible involvement of TLR4 in Dox-induced cardiotoxicity via the implication in cardiomyocyte apoptosis.
4. The TLR4 Expression in Dox-Induced in Cardiomyocytes: Reports from In Vivo Studies
Several studies have demonstrated that Dox treatment leads to impaired cardiac function in rodents, as assessed via echocardiography and invasive hemodynamic assessment [14][23][40]. Cellular and molecular studies have also been carried out using the cardiomyocytes isolated from these Dox-treated animals, and the results are largely consistent with the findings from the in vitro models discussed in the previous section. Previous in vivo studies have shown that Dox enhanced the expression of TLR4 in the cardiac tissue of rodents [14][23][40]. Furthermore, the levels of TLR4 ligands, such as HMGB1 and Hsp70, were also significantly increased in the cardiac tissue of Dox-treated mice [40]. This result further confirms that the TLR4 signaling pathway was activated following Dox treatment, leading to the triggering of NF-κB activity, which leads to the generation of pro-inflammatory cytokines in cardiac tissue, as evidenced by increases in TNF-α, IL-6, IL-13, monocyte chemotactic protein (MCP-1), and transforming growth factor (TGF)-β1 [14][40]. Consistently, the expression of cardiac macrophage markers, including CD45 and CD68, was also elevated in Dox-treated mice [14]. Taken together, elevated cardiac macrophages expressing TLR4 can lead to overwhelming pro-inflammatory cytokines in Dox-treated animals [14][40]. In addition to cardiac inflammation, cardiac remodeling was also observed in Dox-treated mice [40], which was attributed to the fact that TGF-β1 was increased in the hearts of these mice [40]. TGF-β1 is involved in the cardiac remodeling process, since it is a multifunctional cytokine and a growth factor that plays multiple roles in inflammation and fibrosis [41]. Therefore, an increase in cardiac TGF-β1 expression promoted collagen accumulation in the heart of Dox-treated mice, as indicated by increasing α-smooth muscle actin (α-SMA) [40].
Dox induces ROS through redox reactions due to its quinone component, with the resulting production of the superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (∙OH) [8]. This ROS can lead to lipid peroxidation, as indicated by an increase in malondialdehyde (MDA) that was observed in the hearts of Dox-treated rats [14]. In addition, this ROS can trigger apoptosis in cardiac tissue in Dox-treated mice, as evidenced by an increase in Bax, cytochrome c, and TUNEL+ cells, as well as a decrease in Bcl-2 in cardiac tissue [14]. According to in vitro studies [31][32], an in vivo study has also shown that increasing TLR4 activation was implicated in cardiac apoptosis in Dox-treated mice [14]. This could be due to the oxidative stress associated with the increase in TLR4 expression that further promotes inflammation [42], which, in turn, contributes to apoptosis. Therefore, oxidative stress not only directly induces cardiac apoptosis per se but also promotes inflammation via increasing TLR4 expression, leading to cardiac apoptosis in Dox-treated mice.
References
- Anand, P.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116.
- Behranvand, N.; Nasri, F.; Zolfaghari Emameh, R.; Khani, P.; Hosseini, A.; Garssen, J.; Falak, R. Chemotherapy: A double-edged sword in cancer treatment. Cancer Immunol. Immunother. 2022, 71, 507–526.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33.
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2010, 31, 100–110.
- Sohail, M.; Sun, Z.; Li, Y.; Gu, X.; Xu, H. Research progress in strategies to improve the efficacy and safety of doxorubicin for cancer chemotherapy. Expert Rev. Anticancer Ther. 2021, 21, 1385–1398.
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339.
- Benjanuwattra, J.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Doxorubicin and its proarrhythmic effects: A comprehensive review of the evidence from experimental and clinical studies. Pharmacol. Res. 2020, 151, 104542.
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170.
- Wang, L.; Chen, Q.; Qi, H.; Wang, C.; Wang, C.; Zhang, J.; Dong, L. Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage. Cancer Res. 2016, 76, 6631–6642.
- Yarmohammadi, F.; Karbasforooshan, H.; Hayes, A.W.; Karimi, G. Inflammation suppression in doxorubicin-induced cardiotoxicity: Natural compounds as therapeutic options. Naunyn-Schmiedebergs Arch. Pharmacol. 2021, 394, 2003–2011.
- Hadi, N.; Yousif, N.G.; Al-amran, F.G.; Huntei, N.K.; Mohammad, B.I.; Ali, S.J. Vitamin E and telmisartan attenuates doxorubicin induced cardiac injury in rat through down regulation of inflammatory response. BMC Cardiovasc. Disord. 2012, 12, 63.
- Singla, D.K.; Johnson, T.A.; Tavakoli Dargani, Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224.
- Lan, Y.; Wang, Y.; Huang, K.; Zeng, Q. Heat Shock Protein 22 Attenuates Doxorubicin-Induced Cardiotoxicity via Regulating Inflammation and Apoptosis. Front. Pharmacol. 2020, 11, 257.
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752.
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412.
- Spirig, R.; Tsui, J.; Shaw, S. The Emerging Role of TLR and Innate Immunity in Cardiovascular Disease. Cardiol. Res. Pract. 2012, 2012, 181394.
- Sumneang, N.; Oo, T.T.; Singhanat, K.; Maneechote, C.; Arunsak, B.; Nawara, W.; Pratchayasakul, W.; Benjanuwattra, J.; Apaijai, N.; Liang, G.; et al. Inhibition of myeloid differentiation factor 2 attenuates cardiometabolic impairments via reducing cardiac mitochondrial dysfunction, inflammation, apoptosis and ferroptosis in prediabetic rats. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2022, 1868, 166301.
- Oo, T.T.; Sumneang, N.; Ongnok, B.; Arunsak, B.; Chunchai, T.; Kerdphoo, S.; Apaijai, N.; Pratchayasakul, W.; Liang, G.; Chattipakorn, N.; et al. L6H21 protects against cognitive impairment and brain pathologies via toll-like receptor 4–myeloid differentiation factor 2 signalling in prediabetic rats. Br. J. Pharmacol. 2022, 179, 1220–1236.
- Alanazi, A.M.; Fadda, L.; Alhusaini, A.; Ahmad, R.; Hasan, I.H.; Mahmoud, A.M. Liposomal Resveratrol and/or Carvedilol Attenuate Doxorubicin-Induced Cardiotoxicity by Modulating Inflammation, Oxidative Stress and S100A1 in Rats. Antioxidants 2020, 9, 159.
- Sumneang, N.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Myeloid differentiation factor 2 in the heart: Bench to bedside evidence for potential clinical benefits? Pharmacol. Res. 2021, 163, 105239.
- Riad, A.; Bien, S.; Gratz, M.; Escher, F.; Westermann, D.; Heimesaat, M.M.; Bereswill, S.; Krieg, T.; Felix, S.B.; Schultheiss, H.P.; et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 2008, 10, 233–243.
- Baniahmad, B.; Safaeian, L.; Vaseghi, G.; Rabbani, M.; Mohammadi, B. Cardioprotective effect of vanillic acid against doxorubicin-induced cardiotoxicity in rat. Res. Pharm. Sci. 2020, 15, 87–96.
- Ahmed, O.M.; Galaly, S.R.; Mostafa, M.M.A.; Eed, E.M.; Ali, T.M.; Fahmy, A.M.; Zaky, M.Y. Thyme Oil and Thymol Counter Doxorubicin-Induced Hepatotoxicity via Modulation of Inflammation, Apoptosis, and Oxidative Stress. Oxid. Med. Cell. Longev. 2022, 2022, 6702773.
- Oo, T.T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Emerging roles of toll-like receptor 4 in chemotherapy-induced neurotoxicity. Neurotoxicology 2022, 93, 112–127.
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708.
- Feng, P.; Yang, Y.; Liu, N.; Wang, S. Baicalin regulates TLR4/IκBα/NFκB signaling pathway to alleviate inflammation in Doxorubicin related cardiotoxicity. Biochem. Biophys. Res. Commun. 2022, 637, 1–8.
- Kashani, B.; Zandi, Z.; Pourbagheri-Sigaroodi, A.; Bashash, D.; Ghaffari, S.H. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J. Cell. Physiol. 2021, 236, 4121–4137.
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316.
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56.
- Yao, Y.; Xu, X.; Zhang, G.; Zhang, Y.; Qian, W.; Rui, T. Role of HMGB1 in doxorubicin-induced myocardial apoptosis and its regulation pathway. Basic Res. Cardiol. 2012, 107, 267.
- Li, B.; Cai, X.; Wang, Y.; Zhu, H.; Zhang, P.; Jiang, P.; Yang, X.; Sun, J.; Hong, L.; Shao, L. Circ-SKA3 Enhances Doxorubicin Toxicity in AC16 Cells Through miR-1303/TLR4 Axis. Int. Heart J. 2021, 62, 1112–1123.
- Xu, L.; Wang, C.; Zou, Z.; Wu, Z. Ozone Attenuated H9c2 Cell Injury Induced by Doxorubicin. J. Cardiovasc. Pharmacol. 2021, 78, e86–e93.
- Tavakoli Dargani, Z.; Singla, D.K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H460–H471.
- He, M.; Bianchi, M.E.; Coleman, T.R.; Tracey, K.J.; Al-Abed, Y. Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol. Med. 2018, 24, 21.
- Tripathi, A.; Shrinet, K.; Kumar, A. HMGB1 protein as a novel target for cancer. Toxicol. Rep. 2019, 6, 253–261.
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484.
- Syukri, A.; Budu; Hatta, M.; Amir, M.; Rohman, M.S.; Mappangara, I.; Kaelan, C.; Wahyuni, S.; Bukhari, A.; Junita, A.R.; et al. Doxorubicin induced immune abnormalities and inflammatory responses via HMGB1, HIF1-α and VEGF pathway in progressive of cardiovascular damage. Ann. Med. Surg. 2022, 76, 103501.
- Beji, S.; D’Agostino, M.; Gambini, E.; Sileno, S.; Scopece, A.; Vinci, M.C.; Milano, G.; Melillo, G.; Napolitano, M.; Pompilio, G.; et al. Doxorubicin induces an alarmin-like TLR4-dependent autocrine/paracrine action of Nucleophosmin in human cardiac mesenchymal progenitor cells. BMC Biol. 2021, 19, 124.
- Ma, Y.; Zhang, X.; Bao, H.; Mi, S.; Cai, W.; Yan, H.; Wang, Q.; Wang, Z.; Yan, J.; Fan, G.-C.; et al. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS ONE 2012, 7, e40763.
- Saadat, S.; Noureddini, M.; Mahjoubin-Tehran, M.; Nazemi, S.; Shojaie, L.; Aschner, M.; Maleki, B.; Abbasi-kolli, M.; Rajabi Moghadam, H.; Alani, B.; et al. Pivotal Role of TGF-β/Smad Signaling in Cardiac Fibrosis: Non-coding RNAs as Effectual Players. Front. Cardiovasc. Med. 2021, 7, 588347.
- Tawadros, P.S.; Powers, K.A.; Ailenberg, M.; Birch, S.E.; Marshall, J.C.; Szaszi, K.; Kapus, A.; Rotstein, O.D. Oxidative Stress Increases Surface Toll-Like Receptor 4 Expression in Murine Macrophages Via Ceramide Generation. Shock 2015, 44, 157–165.
More
Information
Subjects:
Toxicology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
725
Revisions:
2 times
(View History)
Update Date:
25 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No