Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giuseppe Miserocchi | -- | 1338 | 2023-06-16 05:22:58 | | | |
| 2 | Peter Tang | Meta information modification | 1338 | 2023-06-18 15:33:03 | | | | |
| 3 | Giuseppe Miserocchi | Meta information modification | 1338 | 2023-06-19 15:47:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Miserocchi, G. Early Cell Signaling in Developing Lung Edema. Encyclopedia. Available online: https://encyclopedia.pub/entry/45696 (accessed on 24 July 2026).
Miserocchi G. Early Cell Signaling in Developing Lung Edema. Encyclopedia. Available at: https://encyclopedia.pub/entry/45696. Accessed July 24, 2026.
Miserocchi, Giuseppe. "Early Cell Signaling in Developing Lung Edema" Encyclopedia, https://encyclopedia.pub/entry/45696 (accessed July 24, 2026).
Miserocchi, G. (2023, June 16). Early Cell Signaling in Developing Lung Edema. In Encyclopedia. https://encyclopedia.pub/entry/45696
Miserocchi, Giuseppe. "Early Cell Signaling in Developing Lung Edema." Encyclopedia. Web. 16 June, 2023.
Copy Citation
The lung promptly responds to edemagenic conditions through functional adaptations that contrast the increase in microvascular filtration. In hypoxia, thinning of endothelial cells, a decrease in caveolae and AQP-1, and an increase in lipid rafts are observed. The interpretation of this response is that it favors oxygen diffusion and hinders trans-cellular water fluxes. In hydraulic edema, which generates greater capillary water leakages, an increase in cell volume and opposite changes in membrane rafts were observed; further, the remarkable increase in caveolae suggests a potential abluminal–luminal vesicular-dependent fluid reabsorption.
air–blood barrier
plasma membrane
lipid rafts
caveolae
hypoxia
1. Introduction
Oxygen uptake in the lung is assured by an oversized architecture of the air–blood barrier that is essentially based on two geometrical features: a surface area approaching 100 m2 and a thickness as low as 0.5 µm [1]. Further, on morpho-functional ground, the macromolecular assembly of the interstitial compartment, separating the endothelial from the epithelial cell, enables the optimization of oxygen diffusion and, at the same time, prevents any fluid leakages from the blood capillaries [2]. These features reflect the macromolecular structure of the interstitial compartment that separates the endothelial from the epithelial cells. All inflammatory states, of either infectious (bacterial/viral) or sterile types (hypoxia exposure, surgery, and fluid overload), threaten the structural integrity of the interstitial macromolecular matrix by triggering the inflammatory cascade and, as a consequence, may cause fluid leakages from the capillary. The ensuing fluid accumulation in the interstitial compartment and in the alveoli, depending on the severity of the damage, hinders the process of oxygen uptake. This entry is an up-to-date research focusing on the early role of the endothelial cellular signaling response occurring at the level of the air–blood barrier and aiming to shield the lung when facing the risk of developing edema. Two models of edema are considered: hypoxia exposure and fluid overload. The two models differ considering the damage induced on the macromolecular structure of the pulmonary interstitial compartment: hypoxia leads to an increase in water and solute permeability of the capillary barrier, while fluid overload mostly leads to a loss of the mechanical rigidity and compactness of the interstitial compartment. The two edema models actually share a common trait, namely the remarkable increase in the hydraulic pressure of the interstitial fluid resulting from the initial water leak from the capillaries. Accordingly, in both edema models, endothelial and epithelial cells are exposed to time-dependent mechanical stress. Evidence is provided for the difference in the signaling process eliciting a functional response aimed at buffering the specific initial perturbation in the lung water balance caused by either type of edema. Finally, when scaling up from cells to humans, functional considerations are put forward to link the signaling process with the inter-individual differences in vulnerability to lung edema.
2. The Air–Blood Barrier in Physiological Conditions
The terminal lung units include the “extra-alveolar space” and the “true alveolar space” [3], the latter only including the thin portion of the air–blood barrier (0.5 µm thick) devoted to gas exchange. At the level of the air–blood barrier, a dense package of molecules from the proteoglycan family (PG) guarantees impermeability to water and also provides mechanical stability to the lung parenchyma. PGs consist of a core protein with one or more covalently attached glycosaminoglycan (GAG) chains that are highly hydrophilic. The PG family comprises the large chondroitin sulphate sub-family (>1000 kDa) that provides stability to the collagen–elastin network component by filling the voids between these fibrillar molecules [4][5]. Further, the heparan sulphate PG sub-family (300–500 kDa) that extends to the intercellular clefts assures low microvascular permeability to water and proteins [3]. All PGs act as link proteins with other molecules, as well as with the cell surface through low-energy ionic and/or non-covalent bonds; the nature of these bonds allows mobility between structures and avoids shear stresses during lung movements. PGs represent a powerful shield against edema via specific physico-chemical features: (1) being highly hydrophilic, they can capture free water to form gel; (2) the increase in steric hindrance of the gel, coupled with the remarkable rigidity of the macromolecular structure, results in an increase in the hydraulic interstitial pressure from the physiological value −10 cmH2O [6] up to ~+5 cmH2O [7]. This increase remarkably offsets further microvascular filtration. At this stage, named interstitial lung edema, the increase in extravascular water is <10% [7]. A strict control on extravascular water is required as at the level of the air–blood barrier, there is scanty presence of lymphatics [8]. Fluid accumulation in the interstitial compartment would cause an increase in the intermolecular distance at PG’s binding sites; this, in turn, would decrease the intermolecular attraction forces according to the law of the square of the distance in the binding site. Moreover, sustained edemagenic conditions, proceeding along an inflammatory model, lead to the production of reactive oxygen species and activation of metalloproteases that ultimately cause remarkable fragmentation of PGs [9][10][11][12]. Fluid overload was generated by a slow rate of saline infusion (0.5 mL/(kg·min) [10], while hypoxic edema was obtained by exposure to 12% hypoxia for 3 h [12]. The two models differ in the sequence of matrix PG fragmentation in the air–blood barrier. Saline infusion mainly caused the fragmentation of large chondroitin sulphate PGs leading to an increase in tissue mechanical compliance [10]; conversely, in the hypoxia model, the main damage involved the heparan sulphate PGs leading to an increase in water and solute permeability [12]. When damaged PG molecules attain 60% control [12], fluid leakages increase remarkably, and severe edema develops with a time constant of a few minutes [13][14].
3. Early Cell Signaling in the Lung in Response to Developing Edema
Attention was focused on specialized sites of the plasma membranes that may be considered “mobile signaling platforms”, referred to as membrane rafts (MRs), that include lipid rafts and caveolae [15][16][17]. The hypothesis was developed that early cellular signaling might be reflected by changes in the plasma membrane’s composition of endothelial and epithelial cells at the level of the air–blood barrier in response to interstitial edema. MRs have a “quasi-crystalline” state and a different composition compared to the rest of the plasma membrane, in particular concerning lipid structure [16][17][18]. Figure 1 shows a possible model of the lung’s cellular response to developing edema [19].
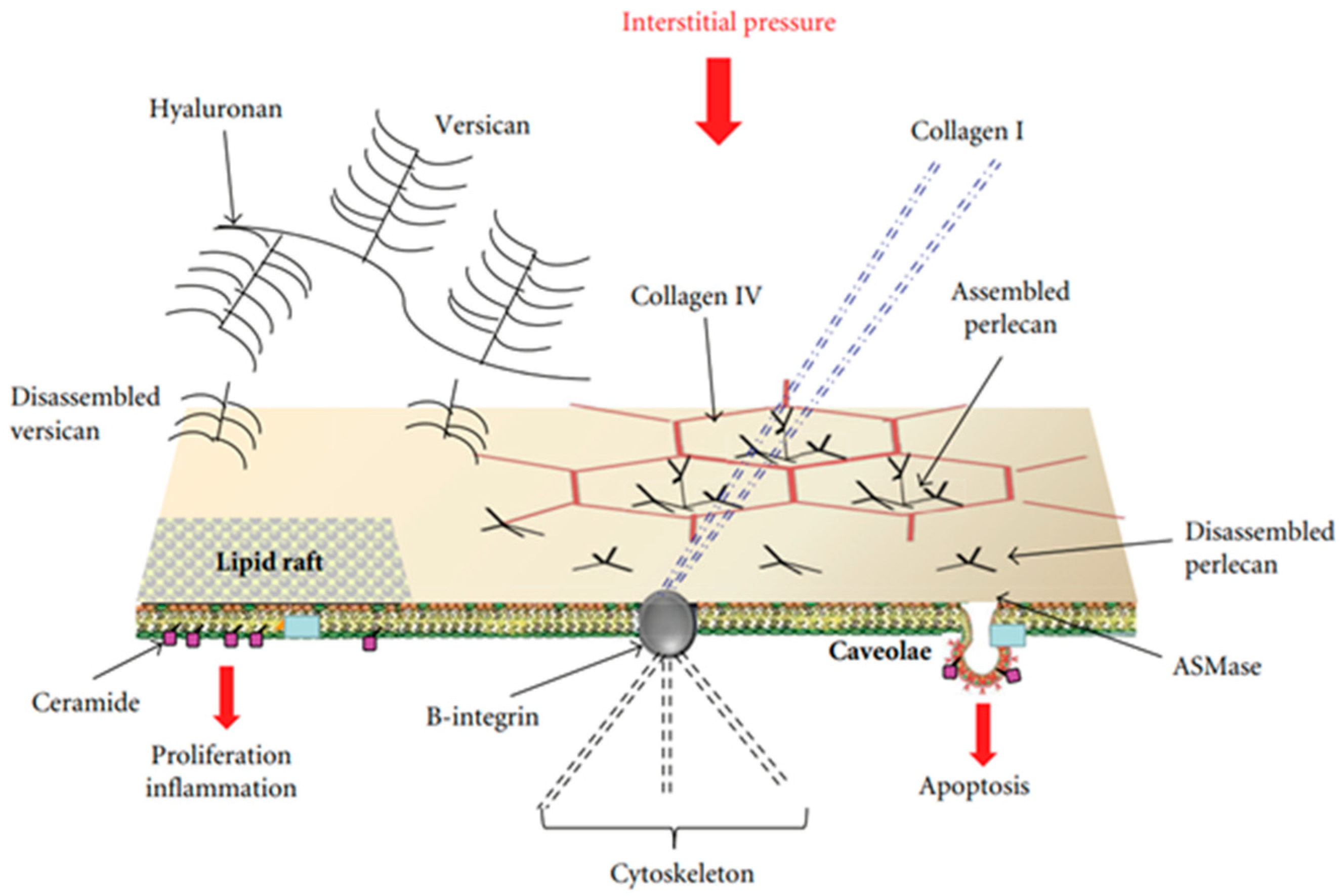
Figure 1. Possible model of lung cellular response to the increase in extravascular water not exceeding 10%. Membrane rafts (MRs), which include lipid rafts and caveolae, are shown. Cell signaling might be activated by mechanical stimulation caused by increased hydraulic pressure of the interstitial fluid through rigid links (collagen I, B-integrin, and cytoskeleton), and/or chemical activation of the MRs by fragments of matrix and basement membrane PGs (hyaluronan, perlecan, and versican). Data from [19].
Lipid rafts are essentially lipid-based domains. Caveolae are flask-shaped invaginations of the plasma membrane with a diameter of ~70 nm (Figure 2); they are protein-based domains due to the presence of caveolin [17][18].

Figure 2. The structure of caveolae and lipid rafts. Data from [19].
Endothelial and epithelial cells are kept flat in a highly deformed state due to their strong attachments to the neighboring cells and the extracellular matrix. Accordingly, their “hard-wired” cytoskeleton might allow them to respond promptly to forces/pressures applied on their surface or changes in cellular volume based on the “tensegrity” concept [20]. Furthermore, the cytoskeleton can contribute to mechano-transduction by transmitting and modulating tension through rigid links involving B-integrin and collagen I as well as focal adhesion with adjacent cells and with the extracellular matrix [21]. MRs are highly movable, dynamic structures that further respond to changes in the concentration of disassembled portions of interstitial macromolecules including PGs (hyaluronan, versican, and perlecan) [22], an event occurring in a sustained edemagenic condition. Finally, the activation of these sites may be stimulated by the increase in water traffic. The hypothesis holds that changes in the lipid composition of the bilayer of the plasma membrane might contribute to triggering the signal transduction process involving MRs [23]. In both edema models, epithelial and endothelial cells are exposed to mechanical forces relating to the remarkable increase in hydraulic pressure of the interstitial fluid. In both models, data were acquired up to 3 h from the onset of the edemagenic condition.
References
- Weibel, E.R. Lung morphometry: The link between structure and function. Cell Tissue Res. 2017, 367, 413–426.
- Beretta, E.; Romanò, F.; Sancini, G.; Grotberg, J.B.; Nieman, G.F.; Miserocchi, G. Pulmonary Interstitial Matrix and Lung Fluid Balance from Normal to the Acutely Injured Lung. Front. Physiol. 2021, 12, 781874.
- Parker, J.C. Hydraulic conductance of lung endothelial phenotypes and Starling safety factors against edema. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2007, 292, L378–L380.
- Cavalcante, F.; Ito, S.; Brewe, K.; Sakai, H.; Alencar, A.; Almeida, M.; Andrade, J.; Majumdar, A.; Ingenito, E.; Suki, B. Mechanical interactions between collagen and proteoglycans: Implications for the stability of lung tissue. J. Appl. Physiol. 2005, 98, 672–679.
- Ma, B.; Bates, J. Mechanical interactions between adjacent airways in the lung. J. Appl. Physiol. 2014, 116, 628–634.
- Miserocchi, G.; Negrini, D.; Gonano, C. Direct measurement of interstitial pulmonary pressure in in situ lung with intact pleural space. J. Appl. Physiol. 1990, 69, 2168–2174.
- Miserocchi, G.; Negrini, D.; Del Fabbro, M.; Venturoli, D. Pulmonary interstitial pressure in intact in-situ lung: Transition to interstitial edema. J. Appl. Physiol. 1993, 74, 1171–1177.
- Schraufnagel, D.E.; Agaram, N.P.; Faruqui, A.; Jain, S.; Jain, L.; Ridg, K.M.; Sznajder, J.I. Pulmonary lymphatics and edema accumulation after brief lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L891–L897.
- Miserocchi, G.; Negrini, D.; Passi, A.; De Luca, G. Development of lung edema: Interstitial fluid dynamics and molecular structure. Physiology 2001, 16, 66–71.
- Negrini, D.; Passi, A.; de Luca, G.; Miserocchi, G. Pulmonary interstitial pressure and proteoglycans during development of pulmonary edema. Am. J. Physiol. 1996, 270 Pt 2, H2000–H2007.
- Passi, A.; Negrini, D.; Albertini, R.; Miserocchi, G.; De Luca, G. The sensitivity of versican from rabbit lung to gelatinase A (MMP2) and B (MMP-9) and its involvement in the development of hydraulic lung edema. FEBS Lett. 1999, 456, 93–96.
- Miserocchi, G.; Passi, A.; Negrini, D.; Del Fabbro, M.; De Luca, G. Pulmonary interstitial pressure and tissue matrix structure in acute hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L881–L887.
- Mazzuca, E.; Aliverti, A.; Miserocchi, G. Computational micro-scale model of control of extravascular water and capillary perfusion in the air blood barrier. J. Theor. Biol. 2016, 400, 42–51.
- Parker, J.C.; Townsley, M.I. Evaluation of lung injury in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L231–L246.
- Allen, J.A.; Halverson-Tamboli, R.; Rasenick, A.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosc. 2007, 8, 128–140.
- Lindner, R.; Naim, H.Y. Domains in biological membranes. Exp. Cell Res. 2009, 315, 2871–2878.
- Lisanti, M.P.; Sherer, P.E.; Vidugiriene, J.; Tang, Z.L.; Hermanowski-Vosatka, A.; Tu, Y.H.; Cook, R.F.; Sargiacomo, M. Characterisation of caveolin rich membrane domains isolated from an endothelial rich source: Implications for human disease. J. Cell Biol. 1994, 126, 111–126.
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007, 8, 185–194.
- Palestini, P.; Botto, L.; Rivolta, I.; Miserocchi, G. Remodelling of membrane rafts expression in lung cells as an early sign of mechanotransduction-signalling in pulmonary edema. J. Lipids 2011, 2011, 695369.
- Ingber, D.E. Tensegrity II. How structural networks influence cellular information processing networks. J. Cell Sci. 2003, 116, 1397–1408.
- Hamill, O.P.; Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 2001, 81, 685–740.
- Palestini, P.; Calvi, C.; Conforti, E.; Daffara, R.; Botto, L.; Miserocchi, G. Compositional changes in lipid microdomains of air-blood barrier plasma membranes in pulmonary interstitial edema. J. Appl. Physiol. 2003, 95, 1446–1452.
- Lundbæk, J.A.; Andersen, O.S.; Werge, T.; Nielsen, C. Cholesterol-induced protein sorting: An analysis of energetic feasibility. Biophys. J. 2003, 84, 2080–2089.
More
Information
Subjects:
Physiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
612
Revisions:
3 times
(View History)
Update Date:
19 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No