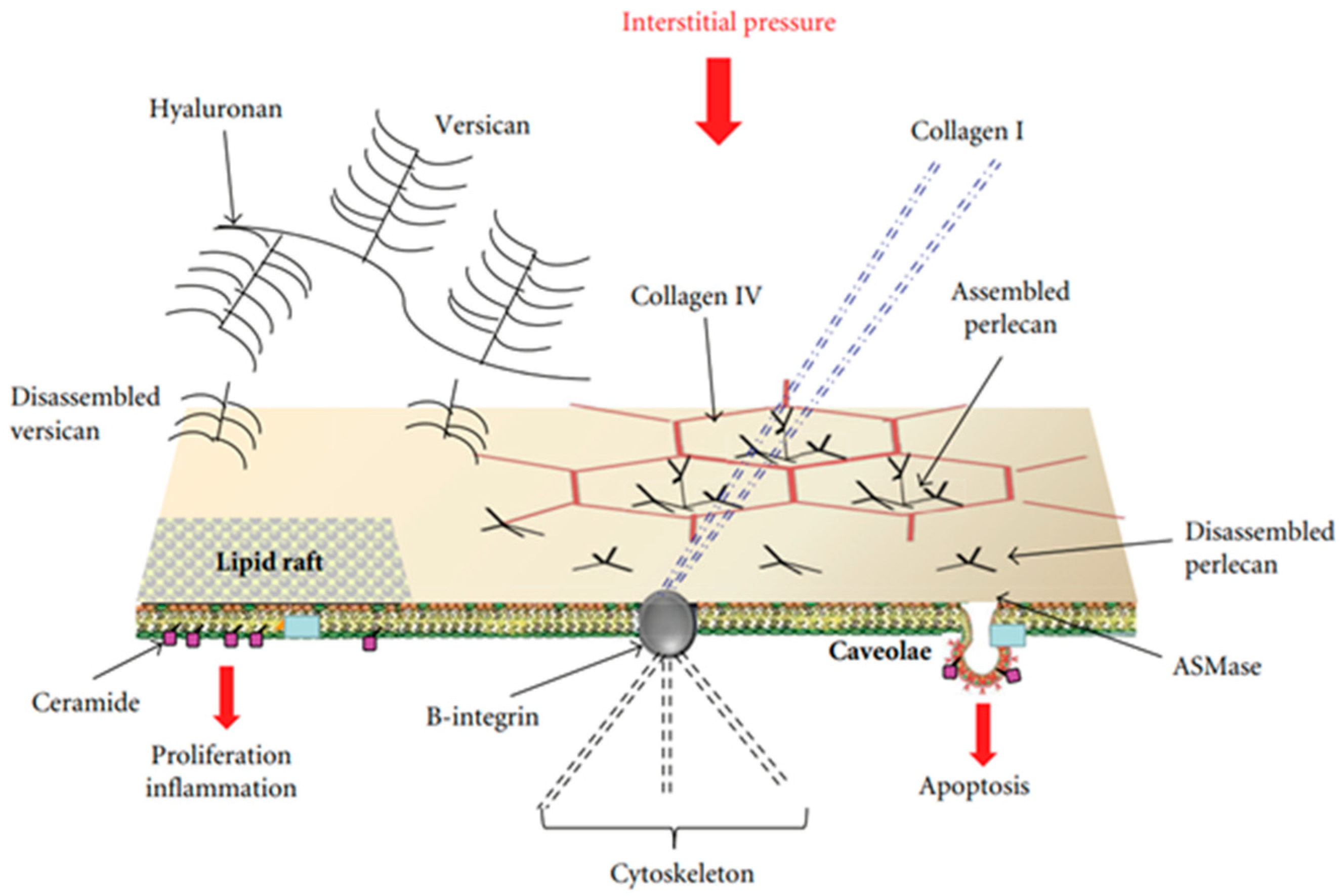
The lung promptly responds to edemagenic conditions through functional adaptations that contrast the increase in microvascular filtration. In hypoxia, thinning of endothelial cells, a decrease in caveolae and AQP-1, and an increase in lipid rafts are observed. The interpretation of this response is that it favors oxygen diffusion and hinders trans-cellular water fluxes. In hydraulic edema, which generates greater capillary water leakages, an increase in cell volume and opposite changes in membrane rafts were observed; further, the remarkable increase in caveolae suggests a potential abluminal–luminal vesicular-dependent fluid reabsorption.
- air–blood barrier
- plasma membrane
- lipid rafts
- caveolae
- hypoxia
1. Introduction
2. The Air–Blood Barrier in Physiological Conditions
3. Early Cell Signaling in the Lung in Response to Developing Edema

This entry is adapted from the peer-reviewed paper 10.3390/life13061240
References
- Weibel, E.R. Lung morphometry: The link between structure and function. Cell Tissue Res. 2017, 367, 413–426.
- Beretta, E.; Romanò, F.; Sancini, G.; Grotberg, J.B.; Nieman, G.F.; Miserocchi, G. Pulmonary Interstitial Matrix and Lung Fluid Balance from Normal to the Acutely Injured Lung. Front. Physiol. 2021, 12, 781874.
- Parker, J.C. Hydraulic conductance of lung endothelial phenotypes and Starling safety factors against edema. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2007, 292, L378–L380.
- Cavalcante, F.; Ito, S.; Brewe, K.; Sakai, H.; Alencar, A.; Almeida, M.; Andrade, J.; Majumdar, A.; Ingenito, E.; Suki, B. Mechanical interactions between collagen and proteoglycans: Implications for the stability of lung tissue. J. Appl. Physiol. 2005, 98, 672–679.
- Ma, B.; Bates, J. Mechanical interactions between adjacent airways in the lung. J. Appl. Physiol. 2014, 116, 628–634.
- Miserocchi, G.; Negrini, D.; Gonano, C. Direct measurement of interstitial pulmonary pressure in in situ lung with intact pleural space. J. Appl. Physiol. 1990, 69, 2168–2174.
- Miserocchi, G.; Negrini, D.; Del Fabbro, M.; Venturoli, D. Pulmonary interstitial pressure in intact in-situ lung: Transition to interstitial edema. J. Appl. Physiol. 1993, 74, 1171–1177.
- Schraufnagel, D.E.; Agaram, N.P.; Faruqui, A.; Jain, S.; Jain, L.; Ridg, K.M.; Sznajder, J.I. Pulmonary lymphatics and edema accumulation after brief lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L891–L897.
- Miserocchi, G.; Negrini, D.; Passi, A.; De Luca, G. Development of lung edema: Interstitial fluid dynamics and molecular structure. Physiology 2001, 16, 66–71.
- Negrini, D.; Passi, A.; de Luca, G.; Miserocchi, G. Pulmonary interstitial pressure and proteoglycans during development of pulmonary edema. Am. J. Physiol. 1996, 270 Pt 2, H2000–H2007.
- Passi, A.; Negrini, D.; Albertini, R.; Miserocchi, G.; De Luca, G. The sensitivity of versican from rabbit lung to gelatinase A (MMP2) and B (MMP-9) and its involvement in the development of hydraulic lung edema. FEBS Lett. 1999, 456, 93–96.
- Miserocchi, G.; Passi, A.; Negrini, D.; Del Fabbro, M.; De Luca, G. Pulmonary interstitial pressure and tissue matrix structure in acute hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L881–L887.
- Mazzuca, E.; Aliverti, A.; Miserocchi, G. Computational micro-scale model of control of extravascular water and capillary perfusion in the air blood barrier. J. Theor. Biol. 2016, 400, 42–51.
- Parker, J.C.; Townsley, M.I. Evaluation of lung injury in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L231–L246.
- Allen, J.A.; Halverson-Tamboli, R.; Rasenick, A.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosc. 2007, 8, 128–140.
- Lindner, R.; Naim, H.Y. Domains in biological membranes. Exp. Cell Res. 2009, 315, 2871–2878.
- Lisanti, M.P.; Sherer, P.E.; Vidugiriene, J.; Tang, Z.L.; Hermanowski-Vosatka, A.; Tu, Y.H.; Cook, R.F.; Sargiacomo, M. Characterisation of caveolin rich membrane domains isolated from an endothelial rich source: Implications for human disease. J. Cell Biol. 1994, 126, 111–126.
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007, 8, 185–194.
- Palestini, P.; Botto, L.; Rivolta, I.; Miserocchi, G. Remodelling of membrane rafts expression in lung cells as an early sign of mechanotransduction-signalling in pulmonary edema. J. Lipids 2011, 2011, 695369.
- Ingber, D.E. Tensegrity II. How structural networks influence cellular information processing networks. J. Cell Sci. 2003, 116, 1397–1408.
- Hamill, O.P.; Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 2001, 81, 685–740.
- Palestini, P.; Calvi, C.; Conforti, E.; Daffara, R.; Botto, L.; Miserocchi, G. Compositional changes in lipid microdomains of air-blood barrier plasma membranes in pulmonary interstitial edema. J. Appl. Physiol. 2003, 95, 1446–1452.
- Lundbæk, J.A.; Andersen, O.S.; Werge, T.; Nielsen, C. Cholesterol-induced protein sorting: An analysis of energetic feasibility. Biophys. J. 2003, 84, 2080–2089.