Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pia Giovannelli | -- | 3052 | 2023-06-14 13:15:42 | | | |
| 2 | Dean Liu | Meta information modification | 3052 | 2023-06-15 02:26:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sabbatino, E.; Tutino, V.; Licitra, F.; Di Donato, M.; Castoria, G.; Migliaccio, A.; Giovannelli, P. Androgen Receptor in Lung Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/45580 (accessed on 23 June 2026).
Sabbatino E, Tutino V, Licitra F, Di Donato M, Castoria G, Migliaccio A, et al. Androgen Receptor in Lung Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/45580. Accessed June 23, 2026.
Sabbatino, Emilia, Viviana Tutino, Fabrizio Licitra, Marzia Di Donato, Gabriella Castoria, Antimo Migliaccio, Pia Giovannelli. "Androgen Receptor in Lung Cancer" Encyclopedia, https://encyclopedia.pub/entry/45580 (accessed June 23, 2026).
Sabbatino, E., Tutino, V., Licitra, F., Di Donato, M., Castoria, G., Migliaccio, A., & Giovannelli, P. (2023, June 14). Androgen Receptor in Lung Cancer. In Encyclopedia. https://encyclopedia.pub/entry/45580
Sabbatino, Emilia, et al. "Androgen Receptor in Lung Cancer." Encyclopedia. Web. 14 June, 2023.
Copy Citation
The androgen receptor (AR) is expressed in many cell types, and its related signaling is widely investigated in hormone-dependent cancers such as prostate and breast. The significance of the AR, however, has been detected even in other cancers, including gastric, bladder, kidney, lung, hepatic, and pancreatic, in which growth and spreading are not strictly or notoriously dependent on sex steroid hormone action.
androgen receptor
gender-related cancers
sex steroid hormones in cancers
1. Introduction
Cancer ranks among the most common causes of death worldwide, and its incidence and mortality are expected to increase due to both the aging of the population and the major diffusion and worsening of some of the risk factors responsible for its onset, such as pollution or an unhealthy lifestyle. In addition to the classic environmental and genomic risk factors, the incidence and mortality of a lot of cancers are also determined by sex or gender. For this reason, an increasing number of scientists have been studying the role of sex steroid hormones in many cancers in addition to reproductive cancers, in which the hormone/hormone receptor action is absolutely the principal guide [1]. To date, different cancers have shown gender disparities, not only in incidence but also in aggressiveness and disease prognosis. Except for breast, thyroid, and other rare cancers located in specific sites of the digestive system, a lot of cancers, such as lung, kidney, bladder, gastric, colorectal, liver, and pancreatic, as well as hepatocarcinoma and many others, show a higher incidence in males [2]. The mechanisms underlying this phenomenon are completely unknown, but there are some clear leading points that can help to understand these cancer-related gender disparities. Occupational risk factors, differences in levels of circulating hormones, and the expressions of their receptors could represent starting points to explain gender disparities in patients with cancers with a higher incidence in males [3]. Even if, between the two sexes, there are no differences in the pivotal mutated genes participating in a cancer’s development, as is the case with the BRAF gene in melanoma or K-RAS in pancreatic cancer, researchers must consider that there are whole groups of genes differentially expressed in response to sex steroid hormones able to influence several processes in cancer. For example, studies analyzing gene expression in clear cell renal carcinoma (ccRCC) have shown that, among the analyzed genes, about 90% were activated in a gender-specific way [3]. Accumulating evidence displays that gender differences also influence the immune system, thereby contributing to the unequal disease outcomes and different efficiency in immune response to therapies in men and women [4].
By reason of the higher incidence in the male gender of many cancers, it is suitable to have a better understanding of the role of sex steroid receptors in gender-related cancers; in particular, it could be advantageous to analyze the role of the androgen receptor (AR) in these gender-related cancers.
AR action has been extensively studied in hormone-dependent cancers such as prostate and breast.
In addition to the classical pathway, the AR can activate the non-classical, or rapid, pathway by an alternative mechanism mediated by different signaling proteins [5][6].
In prostate cancer (PCa), the AR represents a key regulator of tumor development and progression. Several studies in PCa patients treated with anti-androgen or androgen ablation therapy have revealed how androgen/AR signaling mediates many physiological and pathophysiological processes in various tissues/organs [7][8][9][10].
In AR-positive breast cancer, the role of the AR in cell proliferation, apoptosis, migration, and cell invasion is known [5]. Moreover, in triple-negative breast cancer (TNBC), a growing number of studies has clarified the mechanisms used by this receptor to promote cancer progression and aggressiveness [11][12].
In recent years, many researchers have devoted their attention to the actions of the AR in all those cancers not “classically” hormone-dependent, but gender-related, such as lung, kidney, bladder, liver, stomach, and pancreas.
In the group of “classically hormone-dependent cancers” are included all those cancers in which growth and invasiveness are notoriously and directly controlled by sex steroid hormones and their receptors, such as breast cancer in women and prostate cancer in men. Other examples are testicular cancer in men, and uterine and ovarian cancers in women. Except for breast cancer, occurring in both sexes with a clear predominance in women, all these cancers are also sex specific.
Conversely to these, the “non-classically hormone-related cancers”, also known as gender-related cancers, include all those cancers occurring in both sexes, for which it is unknown if any dependence from sex steroid hormones and receptors exists, but there is still an incidence imbalance between men and women. This gap could be explained by the unequal concentrations of circulating hormones between men and women.
2. Androgen Receptor (AR)
The androgen receptor belongs to the large family of type I nuclear receptors. As such, it is a ligand-dependent transcription factor commonly activated by ligand binding. The AR gene is located on the long arm of the X chromosome (Xq11-12) and consists of eight exons that code for the three functional domains typical of steroid hormone receptors: (1) an amino-terminal domain (N-terminal domain, NTD), also indicated as a trans-activation domain (TAD, residues 1–555), (2) a DNA binding domain (DBD, residues 555–623), and (3) a carboxyl-terminal ligand binding domain (LBD, residues 665–919). Finally, a hinge region (residues 623–665) connects the DBD and the LBD [13].
The NTD or TAD represents a variable domain, less conserved than the others. It contains an activation region called AF-1 (ligand-independent transactivation domain) whose absence results in a transcriptional impairment of the receptor’s functions. This region is structurally flexible and is critical in stabilizing the receptor by enhancing the interactions with AR co-activators. The DBD is a highly conserved domain in nuclear receptors; it contributes to androgen receptor dimerization as well as to the binding of specific sequences in chromatin known as androgen response elements (AREs). The LBD turns out to be important in the nuclear localization of the AR. In this domain, there is a region termed AF-2, a ligand-dependent activation region, responsible for the complete activation of the receptor [10].
AR has two isoforms: AR-A and AR-B (Figure 1).
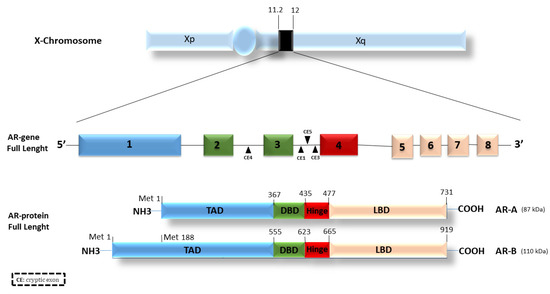
Figure 1. Androgen receptor structure and isoforms. The AR gene is located on the X chromosome and consists of 8 different exons encoding for three distinct functional regions: the TAD (transactivation domain), the DBD (DNA binding domain), and the LBD (ligand binding domain). The DBD and the LBD are linked by a hinge region. Different cryptic exons (CE) are located between exons, i.e., between exons 2, 3, and 4.
The isoform B, also named AR full-length or isoform 1, is widely expressed in most cell types, migrating with an apparent mass of 110 kDa. The transcription of the isoform A starts from the methionine in position 188 (Met-188). The resulting protein migrates with an apparent mass of 87 kDa, lacking 187aa in the N-terminal transactivation domain [14][15]. Both the isoforms are expressed in a sweeping variety of adult and fetal reproductive and non-reproductive tissues [16]. The full-length AR-B represents the predominant AR species in all tissues in which both isoforms have been detected, and, in male and female adult reproductive tissues, AR-B is expressed at high concentrations, whereas AR-A comprises 20% or less of the total AR protein [16]. Furthermore, the ratio of AR-A to AR-B was not shown to change widely in the tissues examined [16]. The two AR isoforms slightly differ in their activity and similarly respond to a variety of androgen agonists and antagonists [17]. The unique differences were studied by Liegibel and colleagues [18]. They proved that AR isoforms have distinct functions in human cells of mesenchymal origin such as osteoblastic cells and genital skin fibroblasts. AR-B was responsible for the mitogenic stimulation of mesenchymal cells, whereas, in AR-positive tissues, AR-A inhibited the mitogenic function of androgen-activated AR-B. AR-A was unable to stimulate cell proliferation, probably due to the reduced binding of AR co-activating protein to the truncated N-terminal TAD [18].
Both the isoforms are expressed in prostate cancer, wherein the AR-B level is still higher than AR-A. Anyway, the AR A/B ratio increases in PCa, in parallel with the Gleason score [15]. These results agree with those obtained in studies of colon cancer, wherein the AR-B expression decreased, whereas the AR-A expression was maintained [19]. The different results in AR-A and -B activity can be explained by considering that all the measurements of their activity were performed using similar levels of the two isoforms, but this does not replicate the normal conditions.
In addition to the classical isoforms, ARs frequently undergo mutations or alternative splicing that causes the formation of alternative splicing variants. Some of these different forms of ARs, represented in Figure 2, can be expressed in normal and cancer tissues and can trigger altered and uncontrolled responses, causing various pathologies or drug resistance in cancer, such as for AR-V3, -V7, or AR-8 in PCa [20][21][22] or for AR-V45 in BC [12]. According to the NCBI site (https://www.ncbi.nlm.nih.gov/gene/367#reference-sequences, accessed on 10 May 2023), there are 5 AR isoforms, indicated as AR 1, 2, 3, 4 and 5 and corresponding to AR-B, AR-V45, AR-V7, AR-V1 and AR-8, respectively.
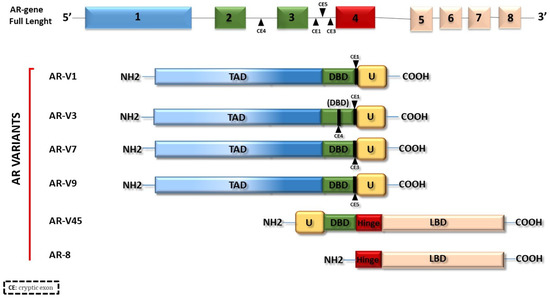
Figure 2. AR variants. The figure illustrates some of the naturally occurring AR variants. Most of them originate from AR alternative splicing.
The presence of the AR and/or its variants makes more complex the molecular scenario of different types of cancers. Studies on the alteration of agonist/antagonist properties of anti-androgens due to different AR mutations have stimulated the search for new drugs that inhibit AR signal transduction [23][24]. It has been seen how mutations in the AR gene and splicing variants can lead, in most cases, to increased cellular aggressiveness in hormone-dependent cancers such as prostate, breast, and ovary [12][25][26][27][28][29][30][31]. All the AR domains can undergo mutations. In particular, the selection pressure of drugs on the AR pathway in PCa increases the number of mutations in the ligand binding domain (LBD), thereby broadening its ligand specificity and sensitivity and reducing the clinical treatment effects of PCa and the quality of patient survival. Although various AR mutations have been reported in prostate cancer, specific hot spot mutations (L702H, W742L/C, H875Y, F877L, and T878A/S) were frequently identified after the gain of drug resistance [32]. A conspicuous group of AR splicing variants, such as AR-V1, 3, 7 and 9, lacks the LBD while showing an intact NTD and DBD and, consequently, a constitutive activity [33][34][35]. Other variants such as AR-V45 and AR-8 lack a DBD and do not work as transcription factors but play different roles. AR-8 promotes cell survival via a non-genomic mechanism [36][37]. AR-V45 is an NTD truncated form, unable to transactivate AR but able to work as a dominant negative and suppress the AR FL functions [37]. The hormone-activated AR works through a genomic or classical pathway and a non-genomic or rapid mechanism. In the genomic mechanism, the AR dimerizes after the binding to its hormone and binds AREs in the promoter region of target genes involved in cell proliferation, epithelial to mesenchymal transitions (EMT), apoptosis, and metabolism [38]. On the other side, the activated AR is also able to influence biological processes, triggering the activation of signaling pathways in few seconds or minutes. These two modes of action are not unconnected, and they work together in same target cells [39], thereby promoting the growth and development of hormone-dependent tumors [40].
The role of the AR has been widely studied in prostate cancer. The AR controls cancer progression and its action is inhibited by reducing androgen synthesis or using specific AR antagonists. However, resistance to these treatments often occurs within 2–3 years of therapy, when patients develop castration-resistant prostate cancer (CRPC), in which active ARs remain as key regulators. Some studies have focused on the functional domains of the AR and its crucial role in this pathology. Deeper knowledge of the structures of the DBD and LBD of the AR provides a framework for understanding the functions of this receptor and can lead to the design of drugs for the treatment of prostate cancer [10].
A great deal of scientific evidence shows the involvement of the AR in female cancers such as ovarian and breast.
Although the molecular mechanisms underlying the androgen receptor role in ovarian cancer are still far from being fully understood, therapeutic approaches designed to modulate androgen receptor activity induce a decrease in tumor progression [27].
In breast cancer (BC), the AR seems to play opposite roles, and this could be attributed to its crosstalk with other signaling pathways. Anyway, whereas in estrogen receptor (ER)-positive breast cancer, the AR appears to both inhibit and promote tumor progression [5][41], in ER-negative BC, particularly in triple-negative breast cancer (TNBC), it mainly promotes tumor progression and tumor growth [5].
It is of paramount importance to increase the understanding of whether the AR signaling pathway also influences the oncogenesis, or the growth and spreading, of gender-related tumors, in order to use targeted strategies to slow tumor progression.
3. AR in Lung Cancer
Lung cancer is the leading cause of cancer death in the world (http://globocan.iarc.fr/, accessed on 10 February 2023). The high mortality is due to late diagnosis, because of the paucity of symptoms in the early stages of this cancer.
Risk factors commonly associated with lung cancer are, above all, tobacco consumption, but also include occupational exposure to cancer-causing agents such as asbestos, radon, and air pollution [42].
Furthermore, the disease shows sex and gender differences, with a higher incidence in men than women [43] and with sex ratios of men to women varying from 1.5 to 2.0 to 1 (http://globocan.iarc.fr, accessed on 10 February 2023). Females tend to be diagnosed younger, at earlier stages and, mostly, with a better prognosis [44]. Worldwide lung cancer mortality is around threefold higher in males, with a current downward trend for males and upward trend for females [44][45]. This trend will slightly change in 2023 in the United States, where the lung cancer incidence will be slightly higher in women than in men, inversely to mortality, which will remain higher in men [45]. Additionally, female patients show better survival rates than males at any stage of disease [46]. Overall, men tend to be less vulnerable to tobacco carcinogens than women [47]. These differences may be due to the increased expression of AR in lung cancer cells [16].
The scientific evidence shows that the adult lung is a target tissue for ARs and suggests that the AR plays a role in lung cancer biology. Mikkonen and colleagues observed not only that the AR is expressed in different human lung cancer types, but also that its expression increases, after androgen treatment, in murine lungs, above all in type II pneumocytes and the bronchial epithelium [48]. Furthermore, androgen treatment significantly alters the gene expression profile in the murine lung and in A549-lung-cancer-derived cells by upregulating transcripts involved in oxygen transport and downregulating those responsible for DNA repair and recombination [48].
Some studies highlight how AR signaling in lung cancer influences tumor progression. Recchia and colleagues demonstrated that in non-small cell lung cancer (NSCLC) cells, specifically in A549 cells, AR and EGFR co-work to trigger cell growth. The molecular mechanisms underlying the crosstalk between growth factors and steroid hormones have been studied, mainly in androgen-sensitive PCa LNCaP cells expressing both ARs and EGFRs [49]. The interaction between dihydrotestosterone (DHT) and ARs controls the expression of AR-responsive genes, such as cyclin D1, at both the transcript and protein levels. In A549 cells, the cooperation between ARs and EGFRs activates p38, thereby regulating the CD1-mTOR pathway and lung cancer proliferation and progression [50].
Similar results were obtained by Lu and co-workers, who reported the downregulation of cyclin D1 and the suppression of cell proliferation and anchorage-independent growth after AR-knockdown via AR-siRNA in A549 cells. In these cells, the AR-siRNA reduced the expression level of another protein, OCT4, implicated in tumor progression and metastasis [51].
In addition to cyclin D1 and OCT4, there is a multitude of proteins whose transcription is AR-dependent. Angiopoietin-like 4 (ANGPTL4) is upregulated by androgens in murine lung and is involved in the selective formation of lung metastases under transforming growth factor (TGF) control [52]. TMPRSS2 gene results upregulated upon androgen exposure [48]; it is a target of ARs in the prostate [53] and a locus for translocation of erythroblast transformation specific (ETS) transcription factors in about 50% of prostate cancers [54]. Currently, however, it is not known whether a similar translocation takes place in a subset of lung cancers; indeed, the TMPRSS2–ERG fusion gene seems to be specific to prostate cancer, which may be due to the strong induction of TMPRSS2 by androgen [55]. A series of more than 60,000 cancer cases was used to determine the frequency of the TMPRSS2–ERG fusion, assayed by comprehensive genomic profiling (CGP). The fusion gene was detected exclusively in tumor samples from male patients, 30% of which were classified as prostatic cancers. Furthermore, the TMPRSS2–ERG gene was also identified in four cases of lung cancer, four cases of bladder cancer, and two cases of pancreatic cancer [56]. Unfortunately, this seems to be a lone study demonstrating the presence of the TMPRSS2–ERG fusion gene in non-prostatic cancers. Further analysis might be done to demonstrate if the presence of this AR-related gene can be used in other AR-positive cancers. The studies so far presented highlight the role of the androgen receptor as a negative prognostic factor in the onset of lung cancer; moreover, the AR could be used as a molecular target in cancer subtypes that overexpress it, such as NSCLC. It is important, therefore, to further investigate the involvement of this receptor in lung cancer.
Although lung cancer has long been a disease characterized by late-stage diagnosis and no progress in treatment options, in the last decade, lung cancer screening in high-risk populations has yielded encouraging results and substantial progress has been made with personalized therapies chosen on the basis of cancer subtype and stage.
To bolster this new and effective approach to lung cancer therapy, it is mandatory to discover new molecular targets, and the AR could represent a good candidate. Gockel et al. have exploited the ubiquitin–proteasome system with proteolysis-targeted chimeras (PROTACs) to degrade ARs in lung cancer cells [57]. The PROTACs, based on the use of the anti-androgen enzalutamide, were able to robustly induce AR degradation in lung cancer cells [57]. Considering that the reduction of AR levels represents, for many scientists, a way to fight lung cancer [50][58], this technique could be an innovative and promising therapeutic strategy. A similar approach is currently used in prostate cancer patients with beneficial results [59][60][61].
These findings have stimulated research in investigating new drugs targeting the AR in monotherapies or therapies combined with the currently available antiandrogens in the management of lung cancer patients.
References
- Giovannelli, P.; Ramaraj, P.; Williams, C. Editorial: Role of Sex Steroids and Their Receptor in Cancers. Front. Endocrinol. 2022, 13, 883229.
- Lopes-Ramos, C.M.; Quackenbush, J.; DeMeo, D.L. Genome-Wide Sex and Gender Differences in Cancer. Front. Oncol. 2020, 10, 597788.
- Brannon, A.R.; Haake, S.M.; Hacker, K.E.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Rathmell, W.K. Meta-Analysis of Clear Cell Renal Cell Carcinoma Gene Expression Defines a Variant Subgroup and Identifies Gender Influences on Tumor Biology. Eur. Urol. 2012, 61, 258–268.
- Markle, J.G.; Fish, E.N. SeXX Matters in Immunity. Trends Immunol. 2014, 35, 97–104.
- Giovannelli, P.; Di Donato, M.; Giraldi, T.; Migliaccio, A.; Castoria, G.; Auricchio, F. Targeting Rapid Action of Sex Steroid Receptors in Breast and Prostate Cancers. Front. Biosci. 2011, 16, 2224–2232.
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187.
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular Determinants of Resistance to Antiandrogen Therapy. Nat. Med. 2004, 10, 33–39.
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen Receptor Antagonists for Prostate Cancer Therapy. Endocr. Relat. Cancer 2014, 21, T105–T118.
- Niu, Y.; Chang, T.M.; Yeh, S.; Ma, W.L.; Wang, Y.Z.; Chang, C. Differential Androgen Receptor Signals in Different Cells Explain Why Androgen-Deprivation Therapy of Prostate Cancer Fails. Oncogene 2010, 29, 3593–3604.
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E. Androgen Receptor: Structure, Role in Prostate Cancer and Drug Discovery. Acta Pharmacol. Sin. 2015, 36, 3–23.
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490.
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492.
- Claessens, F.; Denayer, S.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Haelens, A. Diverse Roles of Androgen Receptor (AR) Domains in AR-Mediated Signaling. Nucl. Recept. Signal. 2008, 6, e008.
- Contrò, V.; Basile, J.R.; Proia, P. Sex Steroid Hormone Receptors, Their Ligands, and Nuclear and Non-Nuclear Pathways. AIMS Mol. Sci. 2015, 2, 294–310.
- Zeng, R.; Liu, Z.; Sun, Y.; Xu, C. Differential Expression and Function of AR Isoforms in Prostate Cancer. Oncol. Rep. 2012, 27, 492–498.
- Wilson, C.M.; McPhaul, M.J. A and B Forms of the Androgen Receptor Are Expressed in a Variety of Human Tissues. Mol. Cell. Endocrinol. 1996, 120, 51–57.
- Gao, T.; McPhaul, M.J. Functional Activities of the A and B Forms of the Human Androgen Receptor in Response to Androgen Receptor Agonists and Antagonists. Mol. Endocrinol. 1998, 12, 654–663.
- Liegibel, U.M.; Sommer, U.; Boercsoek, I.; Hilscher, U.; Bierhaus, A.; Schweikert, H.U.; Nawroth, P.; Kasperk, C. Androgen Receptor Isoforms AR-A and AR-B Display Functional Differences in Cultured Human Bone Cells and Genital Skin Fibroblasts. Steroids 2003, 68, 1179–1187.
- Catalano, M.G.; Pfeffer, U.; Raineri, M.; Ferro, P.; Curto, A.; Capuzzi, P.; Corno, F.; Berta, L.; Fortunati, N. Altered Expression of Androgen-Receptor Isoforms in Human Colon-Cancer Tissues. Int. J. Cancer 2000, 86, 325–330.
- Nagandla, H.; Robertson, M.J.; Putluri, V.; Putluri, N.; Coarfa, C.; Weigel, N.L. Isoform-Specific Activities of Androgen Receptor and Its Splice Variants in Prostate Cancer Cells. Endocrinology 2021, 162, bqaa227.
- Dehm, S.M.; Tindall, D.J. Alternatively Spliced Androgen Receptor Variants. Endocr. Relat. Cancer 2011, 18, R183–R196.
- Yang, X.; Guo, Z.; Sun, F.; Li, W.; Alfano, A.; Shimelis, H.; Chen, M.; Brodie, A.M.H.; Chen, H.; Xiao, Z.; et al. Novel Membrane-Associated Androgen Receptor Splice Variant Potentiates Proliferative and Survival Responses in Prostate Cancer Cells. J. Biol. Chem. 2011, 286, 36152–36160.
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel Mutations of Androgen Receptor: A Possible Mechanism of Bicalutamide Withdrawal Syndrome. Cancer Res. 2003, 63, 149–153.
- Yoshida, T.; Kinoshita, H.; Segawa, T.; Nakamura, E.; Inoue, T.; Shimizu, Y.; Kamoto, T.; Ogawa, O. Antiandrogen Bicalutamide Promotes Tumor Growth in a Novel Androgen-Dependent Prostate Cancer Xenograft Model Derived from a Bicalutamide-Treated Patient. Cancer Res. 2005, 65, 9611–9616.
- Culig, Z.; Santer, F.R. Androgen Receptor Signaling in Prostate Cancer. Cancer Metastasis Rev. 2014, 33, 413–427.
- Li, A.J.; McAllister, P.; Karlan, B.Y. Impact of Androgen Receptor Cytosine-Adenine-Guanine Polymorphisms on Clinical Outcome in BRCA Mutation-Associated Epithelial Ovarian Cancers. Gynecol. Oncol. 2010, 116, 105–108.
- Mizushima, T.; Miyamoto, H. The Role of Androgen Receptor Signaling in Ovarian Cancer. Cells 2019, 8, 176.
- Zhu, H.; Zhu, X.; Zheng, L.; Hu, X.; Sun, L.; Zhu, X. The Role of the Androgen Receptor in Ovarian Cancer Carcinogenesis and Its Clinical Implications. Oncotarget 2017, 8, 29395–29405.
- Chung, W.-M.; Chen, L.; Chang, W.-C.; Su, S.-Y.; Hung, Y.-C.; Ma, W.-L. Androgen/Androgen Receptor Signaling in Ovarian Cancer: Molecular Regulation and Therapeutic Potentials. Int. J. Mol. Sci. 2021, 22, 7748.
- Calvillo-Robledo, A.; Pedernera, E.; Morales-Vásquez, F.; Pérez-Montiel, D.; Gómora, M.J.; Almaraz, M.Á.; De Alba Graue, P.G.; Rendón, E.; López-Basave, H.N.; Quintanar-Stephano, A.; et al. Simultaneous Expression of Steroid Sulfatase and Androgen Receptor Reduced Overall Survival of Patients with Epithelial Ovarian Tumors. J. Ovarian Res. 2021, 14, 98.
- Manning-Geist, B.L.; Gordhandas, S.B.; Giri, D.D.; Iasonos, A.; Zhou, Q.; Girshman, J.; O’Cearbhaill, R.E.; Zamarin, D.; Lichtman, S.M.; Sabbatini, P.J.; et al. Phase II Study of Enzalutamide in Androgen Receptor Positive, Recurrent, High- and Low-Grade Serous Ovarian Cancer. Gynecol. Oncol. 2022, 164, 12–17.
- Shiota, M.; Akamatsu, S.; Tsukahara, S.; Nagakawa, S.; Matsumoto, T.; Eto, M. Androgen Receptor Mutations for Precision Medicine in Prostate Cancer. Endocr. Relat. Cancer 2022, 29, R143–R155.
- Konda, P.; Viswanathan, S.R. How Splicing Confers Treatment Resistance in Prostate Cancer. eLife 2022, 11, e82070.
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively Active Androgen Receptor Splice Variants Expressed in Castration-Resistant Prostate Cancer Require Full-Length Androgen Receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765.
- Cao, S.; Zhan, Y.; Dong, Y. Emerging Data on Androgen Receptor Splice Variants in Prostate Cancer. Endocr. Relat. Cancer 2016, 23, T199–T210.
- Lu, C.; Luo, J. Decoding the Androgen Receptor Splice Variants. Transl. Androl. Urol. 2013, 2, 178–186.
- Zhang, H.; Zhan, Y.; Liu, X.; Qi, Y.; Zhang, G.; Sartor, O.; Dong, Y. Splicing Variants of Androgen Receptor in Prostate Cancer. Am. J. Clin. Exp. Urol. 2013, 1, 18–24.
- Di Croce, L.; Okret, S.; Kersten, S.; Gustafsson, J.-Å.; Parker, M.; Wahli, W.; Beato, M. Steroid and Nuclear Receptors Villefranche-Sur-Mer, France, May 25–27, 1999. EMBO J. 1999, 18, 6201–6210.
- Vicent, G.P.; Nacht, A.S.; Zaurín, R.; Ballaré, C.; Clausell, J.; Beato, M. Minireview: Role of Kinases and Chromatin Remodeling in Progesterone Signaling to Chromatin. Mol. Endocrinol. 2010, 24, 2088–2098.
- Castoria, G.; Giovannelli, P.; Lombardi, M.; De Rosa, C.; Giraldi, T.; de Falco, A.; Barone, M.V.; Abbondanza, C.; Migliaccio, A.; Auricchio, F. Tyrosine Phosphorylation of Estradiol Receptor by Src Regulates Its Hormone-Dependent Nuclear Export and Cell Cycle Progression in Breast Cancer Cells. Oncogene 2012, 31, 4868–4877.
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L. Androgen Receptor Inhibits Estrogen Receptor-α Activity and Is Prognostic in Breast Cancer. Cancer Res. 2009, 69, 6131–6140.
- Alberg, A.J.; Brock, M.V.; Ford, J.G.; Samet, J.M.; Spivack, S.D. Epidemiology of Lung Cancer: Diagnosis and Management of Lung Cancer: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2013, 143, e1S–e29S.
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global Patterns of Cancer Incidence and Mortality Rates and TrendsGlobal Patterns of Cancer. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 1893–1907.
- Stabellini, N.; Bruno, D.S.; Dmukauskas, M.; Barda, A.J.; Cao, L.; Shanahan, J.; Waite, K.; Montero, A.J.; Barnholtz-Sloan, J.S. Sex Differences in Lung Cancer Treatment and Outcomes at a Large Hybrid Academic-Community Practice. JTO Clin. Res. Rep. 2022, 3, 100307.
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- Fu, J.B.; Kau, T.Y.; Severson, R.K.; Kalemkerian, G.P. Lung Cancer in Women: Analysis of the National Surveillance, Epidemiology, and End Results Database. Chest 2005, 127, 768–777.
- Investigators, I.E.L.C.A.P. Survival of Patients with Stage I Lung Cancer Detected on CT Screening. N. Engl. J. Med. 2006, 355, 1763–1771.
- Mikkonen, L.; Pihlajamaa, P.; Sahu, B.; Zhang, F.-P.; Jänne, O.A. Androgen Receptor and Androgen-Dependent Gene Expression in Lung. Mol. Cell. Endocrinol. 2010, 317, 14–24.
- Migliaccio, A.; Castoria, G.; DOMENICO, M.D.; Ciociola, A.; Lombardi, M.; De Falco, A.; Nanayakkara, M.; Bottero, D.; De Stasio, R.; Varricchio, L. Crosstalk between EGFR and Extranuclear Steroid Receptors. Ann. N. Y. Acad. Sci. 2006, 1089, 194–200.
- Recchia, A.G.; Musti, A.M.; Lanzino, M.; Panno, M.L.; Turano, E.; Zumpano, R.; Belfiore, A.; Andò, S.; Maggiolini, M. A Cross-Talk between the Androgen Receptor and the Epidermal Growth Factor Receptor Leads to P38MAPK-Dependent Activation of MTOR and CyclinD1 Expression in Prostate and Lung Cancer Cells. Int. J. Biochem. Cell Biol. 2009, 41, 603–614.
- Lu, H.-H.; Yeh, S.-D.; Chou, Y.-T.; Tsai, Y.-T.; Chang, C.; Wu, C.-W. Androgen Receptor Regulates Lung Cancer Progress through Modulation of OCT-4 Expression. Cancer Res. 2011, 71, 2126.
- Padua, D.; Zhang, X.H.-F.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFβ Primes Breast Tumors for Lung Metastasis Seeding through Angiopoietin-like 4. Cell 2008, 133, 66–77.
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Jänne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A Hierarchical Network of Transcription Factors Governs Androgen Receptor-Dependent Prostate Cancer Growth. Mol. Cell 2007, 27, 380–392.
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent Gene Fusions in Prostate Cancer. Nat. Rev. Cancer 2008, 8, 497–511.
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.-M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG Fusion-Associated Deletions Provide Insight into the Heterogeneity of Prostate Cancer. Cancer Res. 2006, 66, 8337–8341.
- Lara, P.N.; Heilmann, A.M.; Elvin, J.A.; Parikh, M.; De Vere White, R.; Gandour-Edwards, R.; Evans, C.P.; Pan, C.-X.; Schrock, A.B.; Erlich, R.; et al. TMPRSS2-ERG Fusions Unexpectedly Identified in Men Initially Diagnosed with Nonprostatic Malignancies. JCO Precis. Oncol. 2017, 1, 1–6.
- Gockel, L.M.; Pfeifer, V.; Baltes, F.; Bachmaier, R.D.; Wagner, K.G.; Bendas, G.; Gütschow, M.; Sosič, I.; Steinebach, C. Design, Synthesis, and Characterization of PROTACs Targeting the Androgen Receptor in Prostate and Lung Cancer Models. Arch. Pharm. 2022, 355, 2100467.
- Yeh, S.-D.; Yang, P.-C.; Lu, H.-H.; Chang, C.; Wu, C.-W. Targeting Androgen Receptor as a New Potential Therapeutic Approach to Battle Tobacco Carcinogens-Induced Non-Small Cell Lung Cancer. J. Transl. Med. 2012, 10, A8.
- Jia, X.; Han, X. Targeting Androgen Receptor Degradation with PROTACs from Bench to Bedside. Biomed. Pharmacother. 2023, 158, 114112.
- PROTAC Shrinks Mutated Prostate Tumors. Cancer Discov. 2022, 12, OF2.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
15 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No