Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jong-Lyel Roh | -- | 2684 | 2023-06-14 10:38:16 | | | |
| 2 | Camila Xu | Meta information modification | 2684 | 2023-06-14 10:44:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lee, J.; Roh, J. Structure and Function of FSP1. Encyclopedia. Available online: https://encyclopedia.pub/entry/45556 (accessed on 23 July 2026).
Lee J, Roh J. Structure and Function of FSP1. Encyclopedia. Available at: https://encyclopedia.pub/entry/45556. Accessed July 23, 2026.
Lee, Jaewang, Jong-Lyel Roh. "Structure and Function of FSP1" Encyclopedia, https://encyclopedia.pub/entry/45556 (accessed July 23, 2026).
Lee, J., & Roh, J. (2023, June 14). Structure and Function of FSP1. In Encyclopedia. https://encyclopedia.pub/entry/45556
Lee, Jaewang and Jong-Lyel Roh. "Structure and Function of FSP1." Encyclopedia. Web. 14 June, 2023.
Copy Citation
Ferroptosis suppressor protein 1 (FSP1), an NAD(P)H-ubiquinone oxidoreductase that reduces ubiquinone to ubiquinol, has emerged as a critical player in the regulation of ferroptosis. FSP1 operates independently of the canonical system xc–/glutathione peroxidase 4 pathway, making it a promising target for inducing ferroptosis in cancer cells and overcoming ferroptosis resistance.
ferroptosis
ferroptosis suppressor protein 1
FSP1 inhibitor
1. Introduction
Ferroptosis is a distinctive form of cell death characterized by the iron-dependent accumulation of lipid peroxides [1]. An oncogenic RAS-selective lethal chemical erastin was found to induce ferroptosis and was instrumental in the initial discovery of this form of cell death in 2012 [2]. While the exact mechanisms of ferroptosis induction are still being studied, it has been suggested that ferroptosis may originate from oxytosis, which is a type of neural cell death triggered by glutamate toxicity that inhibits cystine uptake through the cystine/glutamate antiporter system xc– (xCT), ultimately leading to glutathione (GSH) depletion and oxidative stress [3]. Furthermore, oxytosis has been identified as a novel form of cell death that involves lipoxygenase (LOX) activation [4]. Oxytosis was further characterized as cell death resulting from the loss of glutathione peroxidase 4 (GPX4), which induces lipid peroxidation and requires 12/15-LOX activity and apoptosis-inducing factor mediation [5]. Recently, the apoptosis-inducing factor mitochondria-associated 2 (AIFM2), initially described as a proapoptotic gene, was later renamed ferroptosis suppressor protein 1 (FSP1) and identified as a potent GSH-independent protection system that prevents ferroptotic cell death [6]. Ferroptosis has gained significant attention in the scientific community due to its unique morphological and biochemical features, distinguishing it from other cell death forms. In 2018, ferroptosis was formally recognized as a distinct form of cell death, triggered by intracellular oxidative and iron imbalances [7]. This iron-dependent vulnerability leads to lipid peroxidation, distinguishing ferroptosis from other programmed cell death pathways, such as apoptosis. However, the classification of ferroptosis as a regulated cell death pathway has been a topic of discussion and remains an area of ongoing research.
Ferroptosis is characterized by the accumulation of lipid peroxides and redox-active iron, leading to oxidative damage of cellular membranes and ultimately causing cell death (Figure 1) [1][2]. The intracellular labile iron pool plays a crucial role in ferroptosis. It retains the most chelatable redox-active ferrous iron (Fe2+), which generates highly reactive radicals through the Fenton reaction [8]. Iron-dependent enzymes such as 12/15-LOX, P450 oxidoreductase, and prostaglandin-endoperoxide synthase 2 catalyze the reaction between ferrous iron and polyunsaturated fatty acid (PUFA)-PLs, which leads to excessive lipid peroxidation and, ultimately, causes damage to cellular membranes [9]. The esterification and incorporation of arachidonate to PLs in the cellular membrane by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are required for ferroptosis [10]. Peroxisomes, cytoplasmic organelles with membrane-bound oxidative properties, are critically involved in ferroptosis through the biosynthesis of plasmalogens, also known as ether lipids [11]. The study emphasizes the importance of peroxisome-dependent plasmalogen generation as a vulnerable pool of ether lipids that can undergo oxidative damage and contribute to ferroptosis in various cell types, such as cancer cells, neurons, and cardiomyocytes. However, radical-trapping antioxidant (RTA) systems can inhibit the propagation of lipid peroxidation and protect cells from excessive lipid peroxidation. [12]. GPX4 and system xc– are two key regulators of ferroptosis, which have primary antioxidant functions in detoxifying cellular lipid peroxidation [13].
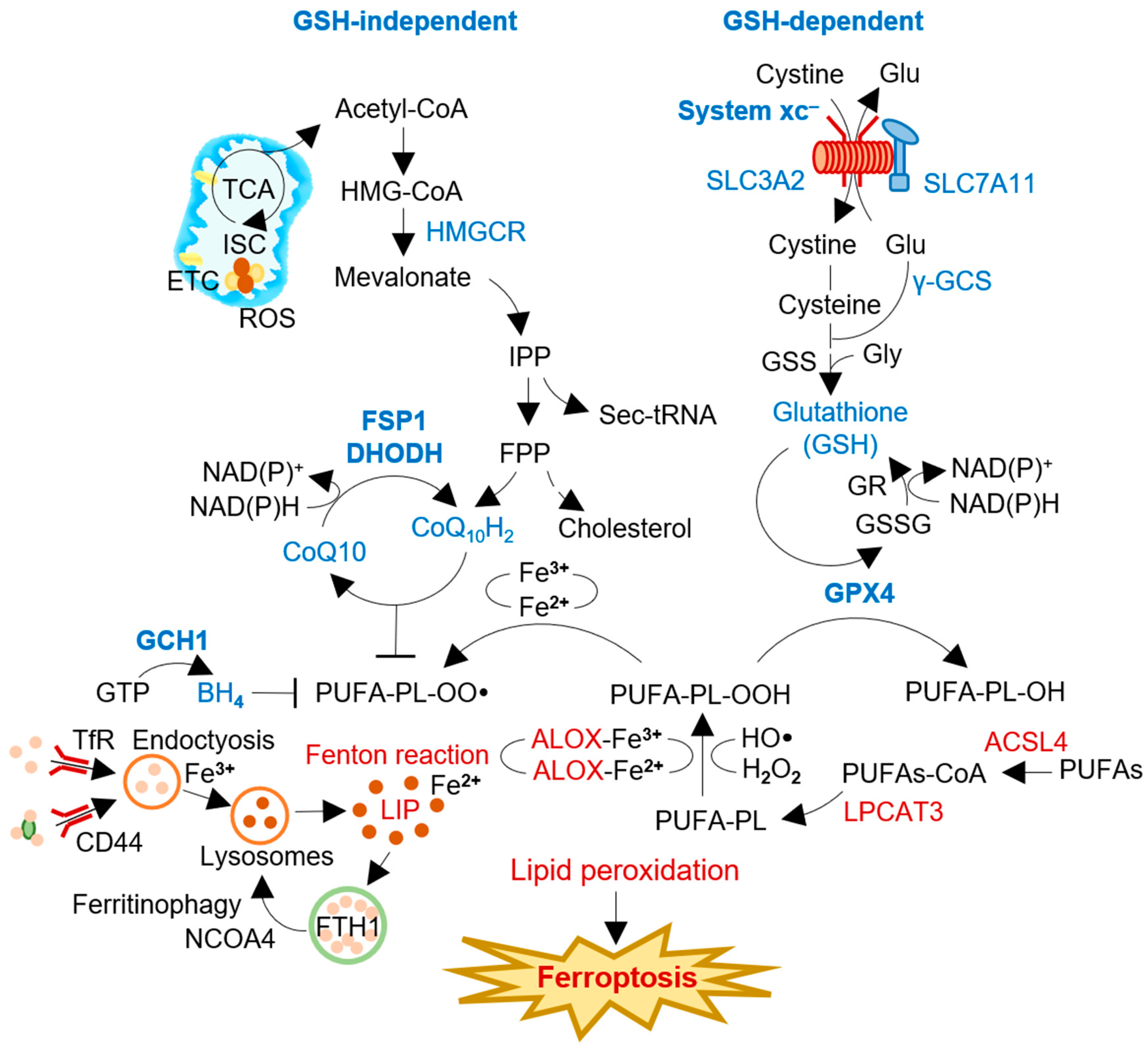
Figure 1. The process and regulators of ferroptosis induction. Intracellular labile iron pool retains chelatable redox-active ferrous iron (Fe2+) that can generate soluble radicals via the Fenton reaction. Radical-trapping antioxidant (RTA) systems can inhibit the propagation of lipid peroxidation and protect cells from excessive lipid peroxidation. System xc– and GPX4 are involved in the GSH-dependent canonical pathway, negatively regulating ferroptosis against membrane lipid peroxidation. FSP1, DHODH, and GCH1 are GSH-independent RTAs using lipophilic radical scavengers of ubiquinol or BH4. FSP1 and DHODH reduced ubiquinone (CoQ10) to ubiquinol (CoQ10H2) with the consumption of NAD(P)H. The regulators inhibit (blue) or promote (red) ferroptosis induction in cancer cells. ACSL4, acyl-CoA synthetase long chain family member 4; ALOX, arachidonate lipoxygenase; BH4, tetrahydrobiopterin; CoA, coenzyme A; DHODH, dihydroorotate dehydrogenase; FPP, farnesyl pyrophosphate; FSP1, ferroptosis suppressor protein 1; GCH1, guanosine triphosphate cyclohydrolase 1; γ-GCS, γ-glutamylcysteine synthetase; Glu, glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; GSS, glutathione synthatase; GSSG, glutathione disulfide; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HO•, hydroxyl radical; IPP, isopentenyl pyrophosphate; ISC, iron-sulfur cluster; LIP, labile iron pool; LPCAT3, lysophosphatidylcholine acyltransferase 3; NADP, nicotinamide adenine dinucleotide phosphate; NCOA4, nuclear receptor coactivator 4; PL-OO•, lipid peroxyl radical; PUFA-PL, polyunsaturated fatty acid-containing phospholipid; PL-PUFA-OH, polyunsaturated fatty acid-containing phospholipid alcohol; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; Sec-tRNA, selenocysteine-tRNA; TCA, tricarboxylic acid cycle; TfR, transferrin receptor; xCT, system xc– cystine/glutamate antiporter.
The involvement of redox-active iron is essential for oxidative damage to membrane lipids in the ferroptosis process [1]. The labile iron pool, a chelatable and redox-active fraction of cellular iron, plays a critical role in cellular iron metabolism [14]. Although a small fraction of the total cellular redox-active iron, cytosolic labile iron plays a crucial role in cellular iron metabolism [15]. Iron homeostasis is regulated through iron uptake via transferrin receptor 1 (TFR1) and its sequestration into ferritins. CD44, a cell surface marker, also facilitates the endocytosis of iron-bound hyaluronates through its interaction with hyaluronates, contributing to iron homeostasis [16]. CD44 contributes to the internalization of both iron and copper. Salinomycin, an antimicrobial agent with demonstrated effects on iron-addicted cancer stem cells, sequesters iron within lysosomes [17][18]. This sequestration leads to a decrease in intracellular iron levels, activating autophagy and resulting in the release of iron through ferritin degradation [19]. The degradation of ferritin during the autophagy process, known as ferritinophagy, is facilitated by nuclear receptor coactivator 4 (NCOA4), selectively enriched in autophagosomes [20]. NCOA4-mediated ferritinophagy plays a critical role in initiating ferroptosis [21]. Furthermore, ferroptosis has been identified as an autophagic cell death process induced by NCOA4-mediated ferritinophagy [22][23].
Preventing ferroptosis by inhibiting lipid peroxyl radicals is not limited to the canonical GPX4 and system xc– antioxidant system. Coenzyme Q10 (CoQ10) can act as a lipid peroxide scavenger and electron transporter [24]. FSP1 and dihydroorotate dehydrogenase (DHODH) are enzymes that reduce CoQ10 (ubiquinone) to CoQ10H2 (ubiquinol) (Figure 1). FSP1 localizes to the plasma membrane through N-terminal myristoylation and can inhibit ferroptosis by functioning as a radical scavenger of lipid peroxides and recycling vitamin E [6][25]. Similarly, DHODH in the inner mitochondrial membrane also contributes to the reduction of CoQ10 [26]. FSP1 and DHODH play a role in preventing ferroptosis, implicating them as promising targets for cancer therapy, particularly in cancers with relatively less functioning of the core ferroptosis regulatory system, xCT/GSH/GPX4 [27]. As a promising target for ferroptosis-based cancer therapy, the depletion of reduced CoQ10 has recently gained attention [28]. Thus, understanding the role of FSP1 in ferroptosis and its modulation, as well as exploring the potential of targeting FSP1 for ferroptosis induction in cancer therapy, is crucial.
2. Understanding the Structure and Function of FSP1
Apoptosis-inducing factors (AIFs) are a group of flavoproteins that can trigger caspase-independent apoptotic cell death [29]. In humans, there are three AIF isozymes: AIFM1, AIFM2, and AIFM3. AIFM1 is the most abundant isozyme and is initially translated into the cytosol before being transported to the mitochondrial membrane [30]. There, it undergoes folding and acquires its functional structure with the help of flavin adenine dinucleotide (FAD). In contrast, AIFM2 (FSP1) lacks a mitochondrial targeting sequence, causing no entrance into the mitochondria and adherence to the outer mitochondrial membrane (OMM), and features an N-terminal myristoylation motif [31]. FSP1 consists of a short hydrophobic region at the N-terminus and a FAD-dependent oxidoreductase domain [32]. Studies have shown that the myristoylation motif is critical for targeting FSP1 to lipid droplets and the plasma membrane, where it interacts with 6-hydroxy-FAD to exert its function (Figure 2) [25]. FSP1 is also associated with mitochondria to induce apoptosis. Deletion mutations at the N-terminal region (aas 1–185 and 1–300) result in nuclear localization and failure to affect cell death [33]. Additionally, domain mapping experiments have revealed that only the C-terminal 187 aa of FSP1 is required for apoptotic induction, whereas mutations in the N-terminal domains responsible for its oxidoreductase function do not affect its apoptotic function [34]. Interestingly, the AIFM2 gene contains a putative p53-binding element in intron 5, suggesting that p53 can activate its gene expression [31][35]. Overall, FSP1 plays an essential role in apoptosis and ferroptosis suppression. Further studies are needed to fully understand its complex regulatory mechanisms and potential as a therapeutic target for cancer treatment.
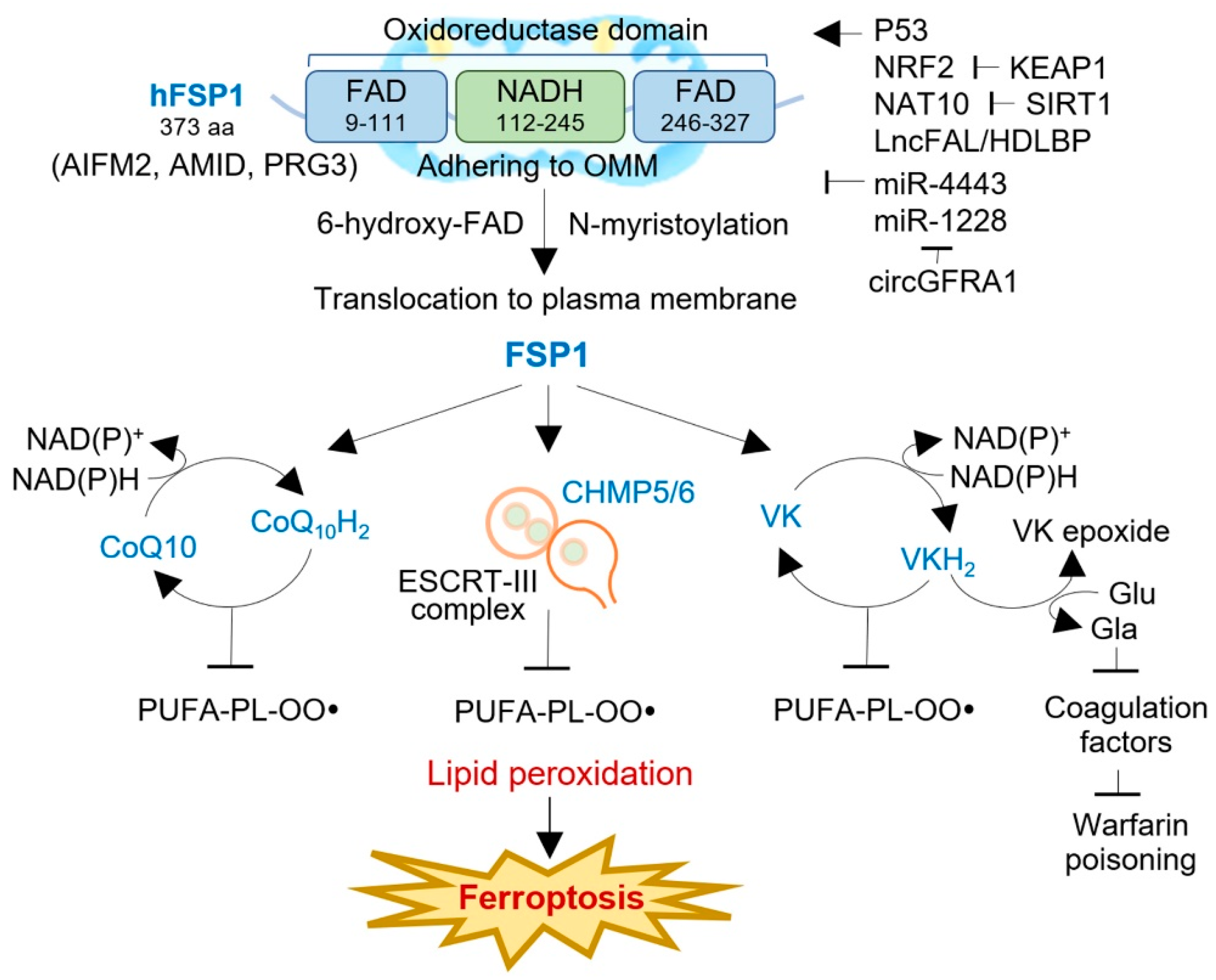
Figure 2. The structure, function, and regulation of FSP1 in human cancers. FSP1 is a NAD(P)H-ubiquinone oxidoreductase, composing FAD- and NADH-dependent domains with an N-terminal myristoylation motif. FSP1 adheres to the outer mitochondrial membrane (OMM) and, after N-myristoylation, is translocated to the plasma membrane or lipid droplets, where it interacts with 6-hydroxyl-FAD. FSP1 exerts ferroptosis resistance via three distinct mechanisms: the FSP1-CoQ10-NAD(P)H pathway, the FSP1-ESCRT-III-dependent membrane repair pathway, and the FSP1-VKH2-NAD(P)H pathway. The activity of FSP1 is regulated by multiple factors, e.g., p53, NRF2, NAT10, LncFAL, miR-4443, mi-1228, and circGFRA1. AIFM2, apoptosis-inducing factor mitochondria-associated 2; AMID, AIF-homologous mitochondrion-associated inducer of death; CHAMP, charged multivesicular body protein; ESCRT, an endosomal sorting complex required for transport; FAD, flavin adenine dinucleotide; Gla, γ-carboxyglutamate; KEAP1, Kelch-like ECH-associated protein 1; NAT10, N-acetyltransferase 10; lncFAL, ferroptosis-associated long non-coding RNA; NRF2, nuclear factor erythroid 2-related factor 2; PRG3, p53-responsive gene 3; SIRT1, sirtuin 1; VK, vitamin K; VKH2, hydroquinone.
FSP1 is a flavoprotein and NAD(P)H-dependent oxidoreductase critical in suppressing ferroptosis. By reducing ubiquinone-10 to ubiquinol-10 at the plasma membrane, using N-myristoylation and NAD(P)H as a substrate, FSP1 inhibits ferroptosis in a GPX4- and GSH-independent manner [6][25]. This reduction of oxidized CoQ10 decreases the pool of oxidized CoQ10, acting as a lipophilic RTA that stops the propagation of lipid peroxides, thus preventing ferroptosis (Figure 2). To confirm the anti-ferroptotic effect of FSP1, co-autoxidation experiments were conducted with egg phosphatidylcholine and STY-BODIPY, which uses a lipophilic alkoxyl radical generator. Additionally, FSP1 can indirectly generate α-tocopherol via CoQ10H2 or directly produce it in vitro, providing a reactive RTA effect [6]. Interestingly, the acute reduction in cellular CoQ levels caused by 4-chlorobenzoic acid (4-CBA) or CoQ2 knockout does not significantly affect the sensitivity to RSL3 as much as FSP1 knockout does, indicating that other FSP1-mediated pathways may contribute to ferroptosis resistance [25]. These findings suggest that FSP1 is a promising therapeutic target for tumors responsive to FSP1 inhibitors.
Researchers conducted a counter-screen experiment on FSP1-overexpressing cells in both wild-type and GPX4 knockout settings, which led to the discovery of a potent FSP1 inhibitor called iFSP1. The strong protective effect of FSP1 on GPX4 knockout cells was the basis for identifying iFSP1 [6]. Previous studies have shown that the first generation of potent FSP1 inhibitors can induce ferroptosis in various tumor cells, and iFSP1 treatment effectively sensitized cancer cells to ferroptosis. In addition, the expression of AIFM2 positively correlates with resistance to GPX4 inhibitors in cancer cell lines, suggesting that FSP1 inhibitors have potential utility as an alternative therapeutic strategy [25]. It is worth noting that the withdrawal of the ferroptosis inhibitor ferrostatin-1 reduced tumor growth in the FSP1/GPX4 double knockout but not the GPX4 single knockout in the H460 lung cancer mouse xenograft model, indicating that targeting FSP1 may be a promising therapeutic strategy to overcome resistance to ferroptotic cell death in these clinical contexts [25].
FSP1 has recently been identified as a crucial suppressor of ferroptotic cell death by reducing the concentration of CoQ10 after treatment with erastin, sorafenib, and RSL3 [36]. Interestingly, the exogenous administration of CoQ10 failed to reverse ferroptotic cell death in FSP1-silenced cells, indicating that other mechanisms are involved in ferroptosis resistance. A recent study has shown that FSP1 regulates the expression of the charged multivesicular body protein 5 (CHMP5) and CHMP6, essential subunits of the endosomal sorting complex required for transport (ESCRT)-III-dependent membrane repair machinery [36]. Knocking down FSP1 suppressed the expression of the CHMP5 and CHMP6 induced by the RSL3 treatment, whereas the overexpression of the CHMP5 restored cell viability and rescued cells from RSL3-, erastin-, and sorafenib-induced cell death in both wild-type and FSP1-silenced cells. These findings suggested that ESCRT-III-dependent membrane repair is another mechanism that underlies FSP1-mediated ferroptosis resistance.
FSP1 functions as a vitamin K reductase, preventing lipid peroxidation by reducing vitamin K to its corresponding hydroquinone (VKH2) [37]. Vitamin K is a redox-active naphthoquinone that resembles ubiquinone, the oxidized form of CoQ [38]. The canonical vitamin K cycle involves the conversion of vitamin K to VKH2, which acts as a potent reactive thiol antioxidant [39]. FSP1 efficiently reduces vitamin K to VKH2, a potent reactive thiol antioxidant that prevents lipid peroxidation [40]. Three types of naturally occurring vitamin K compounds (phylloquinone, menaquinone-4 (MK4), and menadione) protected cells from the ferroptosis induced by GPX4 deletion [37]. All three types of vitamin K also rescued ferroptosis caused by ferroptosis inducers and glutamate-induced neuronal ferroptosis but did not protect against apoptosis, necroptosis, or pyroptosis. FSP1-mediated vitamin K reduction was also responsible for the vitamin K antidotal effect against warfarin poisoning [41]. FSP1 serves as the vitamin K reductase accountable for the warfarin-resistant alternative vitamin K reduction pathway. MK4-treated FSP1−/− mice exhibited a much lower conversion rate of MK4 to MK4 epoxide and significantly prolonged prothrombin time than FSP1+/− mice upon exposure to high doses of warfarin. This result highlighted the critical role of FSP1 in the antidotal effect of high-dose vitamin K against warfarin poisoning [37]. Therefore, FSP1 acts as a vitamin K reductase, producing VKH2 via NAD(P)H consumption to prevent lipid peroxidation, maintain a warfarin-resistant non-canonical vitamin K cycle, and inhibit ferroptosis. However, it is essential to note that FSP1 activity varies among individuals, and individuals with high FSP1 activity may have reduced effectiveness of warfarin. In contrast, those with low FSP1 activity require a higher dose of vitamin K to mitigate the risk of warfarin poisoning.
Previous studies suggest that FSP1, also known as AIFM2 or AMID, may have a significant role in signaling mitochondrial stress. When oxidative stress occurs, FSP1 binds with 4-hydroxy-2-nonenal (HNE), a lipid peroxidation end product, forming a lipid adduct that lacks oxidoreductase activity [42]. The HNE-FSP1 adduct is then transported from the mitochondria to the nucleus, leading to DNA damage and cell death. Notably, doxorubicin treatment can increase cardiac levels of HNE and AIFM2. The HNE adduction of AIFM2 inhibits the NADH oxidoreductase activity of AIFM2, promoting its translocation from mitochondria. This discovery reveals an unexpected role of these proteins in mitochondrial stress signaling and the adverse effects of cancer therapy. Apart from its role in mitochondrial stress signaling, FSP1 can bind to nuclear DNA non-specifically [34]. FSP1 can directly bind to nuclear DNA and alter chromatin condensation. Additionally, FSP1 can induce caspase- and p53-independent apoptosis by disrupting mitochondrial morphology and releasing proapoptotic factors [33]. Under stress conditions, such as hypoxia, which activates p53-mediated apoptosis, FSP1 may stabilize p53 by inhibiting its degradation, accelerating the apoptotic process [35]. Under normal cellular conditions, FSP1 may promote cell survival by generating reactive oxygen species (ROS) to maintain survival signaling [43]. FSP1 is crucial in various cellular processes, including mitochondrial stress signaling, nuclear DNA binding, apoptosis induction, and cell survival. However, its exact mechanisms and functions require further investigation to provide a comprehensive understanding of this protein’s complex actions in various cellular processes.
The discovery of FSP1 as a crucial suppressor of ferroptosis has shed light on the intricate molecular mechanisms underlying this form of cell death. FSP1 exerts its protective effects against ferroptosis. These pathways involve the reduction of CoQ10 and vitamin K, as well as the regulation of ESCRT-III-dependent membrane repair, which collectively contribute to inhibiting lipid peroxidation and preventing ferroptosis. However, several unanswered questions remain regarding the regulation of FSP1 expression and activity. Investigating the molecular structure of FSP1 and elucidating the signaling pathways that control its expression and function would provide valuable insights into the precise mechanisms by which FSP1 suppresses ferroptosis.
Additionally, identifying potential therapeutic targets for developing FSP1 activators or inhibitors holds great promise for developing novel treatment strategies for cancer and other diseases associated with ferroptosis. Further research in these areas has the potential to uncover novel therapeutic approaches that specifically target FSP1 and its related pathways, thus enhancing the understanding of ferroptosis and its implications in various diseases. By exploring the intricate mechanisms of FSP1, researchers can pave the way for developing innovative interventions to modulate ferroptosis and improve patient outcomes.
References
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558.
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506.
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248.
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535.
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98.
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608.
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408.
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849.
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046.
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta 2010, 1800, 783–792.
- Müller, S.; Sindikubwabo, F.; Cañeque, T.; Lafon, A.; Versini, A.; Lombard, B.; Loew, D.; Wu, T.D.; Ginestier, C.; Charafe-Jauffret, E.; et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 2020, 12, 929–938.
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033.
- Versini, A.; Colombeau, L.; Hienzsch, A.; Gaillet, C.; Retailleau, P.; Debieu, S.; Müller, S.; Cañeque, T.; Rodriguez, R. Salinomycin derivatives kill breast cancer stem cells by lysosomal iron targeting. Chemistry 2020, 26, 7416–7424.
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 2011, 31, 2040–2052.
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109.
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv. Exp. Med. Biol. 2021, 1301, 41–57.
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Banerjee, R.; Purhonen, J.; Kallijärvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2022, 289, 6936–6958.
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692.
- Boukalova, S.; Hubackova, S.; Milosevic, M.; Ezrova, Z.; Neuzil, J.; Rohlena, J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165759.
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396.
- Yu, H.; Yan, J.; Li, Z.; Yang, L.; Ju, F.; Sun, Y. Recent trends in emerging strategies for ferroptosis-based cancer therapy. Nanoscale Adv. 2023, 5, 1271–1290.
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684.
- Novo, N.; Ferreira, P.; Medina, M. The apoptosis-inducing factor family: Moonlighting proteins in the crosstalk between mitochondria and nuclei. IUBMB Life 2021, 73, 568–581.
- Wu, M.; Xu, L.G.; Su, T.; Tian, Y.; Zhai, Z.; Shu, H.B. AMID is a p53-inducible gene downregulated in tumors. Oncogene 2004, 23, 6815–6819.
- Vabulas, R.M. Ferroptosis-Related Flavoproteins: Their Function and Stability. Int. J. Mol. Sci. 2021, 22, 430.
- Wu, M.; Xu, L.G.; Li, X.; Zhai, Z.; Shu, H.B. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623.
- Marshall, K.R.; Gong, M.; Wodke, L.; Lamb, J.H.; Jones, D.J.; Farmer, P.B.; Scrutton, N.S.; Munro, A.W. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J. Biol. Chem. 2005, 280, 30735–30740.
- Ohiro, Y.; Garkavtsev, I.; Kobayashi, S.; Sreekumar, K.R.; Nantz, R.; Higashikubo, B.T.; Duffy, S.L.; Higashikubo, R.; Usheva, A.; Gius, D.; et al. A novel p53-inducible apoptogenic gene, PRG3, encodes a homologue of the apoptosis-inducing factor (AIF). FEBS Lett. 2002, 524, 163–171.
- Dai, E.; Zhang, W.; Cong, D.; Kang, R.; Wang, J.; Tang, D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun. 2020, 523, 966–971.
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783.
- Ivanova, D.; Zhelev, Z.; Getsov, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018, 16, 352–358.
- Shearer, M.J.; Okano, T. Key pathways and regulators of vitamin K function and intermediary metabolism. Annu. Rev. Nutr. 2018, 38, 127–151.
- Vervoort, L.M.; Ronden, J.E.; Thijssen, H.H. The potent antioxidant activity of the vitamin K cycle in microsomal lipid peroxidation. Biochem. Pharmacol. 1997, 54, 871–876.
- Jin, D.Y.; Chen, X.; Liu, Y.; Williams, C.M.; Pedersen, L.C.; Stafford, D.W.; Tie, J.K. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat. Commun. 2023, 14, 828.
- Miriyala, S.; Thippakorn, C.; Chaiswing, L.; Xu, Y.; Noel, T.; Tovmasyan, A.; Batinic-Haberle, I.; Vander Kooi, C.W.; Chi, W.; Latif, A.A.; et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 2016, 91, 68–80.
- Gong, M.; Hay, S.; Marshall, K.R.; Munro, A.W.; Scrutton, N.S. DNA binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J. Biol. Chem. 2007, 282, 30331–30340.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.0K
Revisions:
2 times
(View History)
Update Date:
14 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No