Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manabu Kurokawa | -- | 1652 | 2023-06-14 04:54:26 | | | |
| 2 | Jason Zhu | Meta information modification | 1652 | 2023-06-15 05:08:44 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Feng, W.W.; Zuppe, H.T.; Kurokawa, M. Regulation of CD36 Expression and Activity in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/45546 (accessed on 28 July 2026).
Feng WW, Zuppe HT, Kurokawa M. Regulation of CD36 Expression and Activity in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/45546. Accessed July 28, 2026.
Feng, William W., Hannah T. Zuppe, Manabu Kurokawa. "Regulation of CD36 Expression and Activity in Cancer" Encyclopedia, https://encyclopedia.pub/entry/45546 (accessed July 28, 2026).
Feng, W.W., Zuppe, H.T., & Kurokawa, M. (2023, June 14). Regulation of CD36 Expression and Activity in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/45546
Feng, William W., et al. "Regulation of CD36 Expression and Activity in Cancer." Encyclopedia. Web. 14 June, 2023.
Copy Citation
Cluster of differentiation 36 (CD36) is a cell surface scavenger receptor that plays critical roles in many different types of cancer, notably breast, brain, and ovarian cancers. While it is arguably most well-known for its fatty acid uptake functions, it is also involved in regulating cellular adhesion, immune response, and apoptosis depending on the cellular and environmental contexts.
tumor microenvironment
lipid metabolism
cancer stem cells
1. Introduction
The CD36 mRNA codes for a 472-amino-acid-long protein. However, the mature CD36 protein is 471 amino acids long due to loss of the initiator methionine [1]. The majority of the CD36 protein resides in an extracellular region (Gly30-Asn439), anchored to the plasma membrane by two transmembrane domains at the N- and C-terminal ends (Gly8-Val29 and Leu440-Ile461). The extracellular domain contains two large hydrophobic binding pockets, which are essential for the uptake of fatty acids (FAs) and oxidized low-density lipoproteins (oxLDLs). Although only 17 amino acids of CD36 protein are cytoplasmic (Gly2-Cys7 and Ser462-Lys472), these residues are critical for CD36 maturation and signaling activity. As detailed below and in Figure 1, CD36 is a heavily post-translationally modified protein.
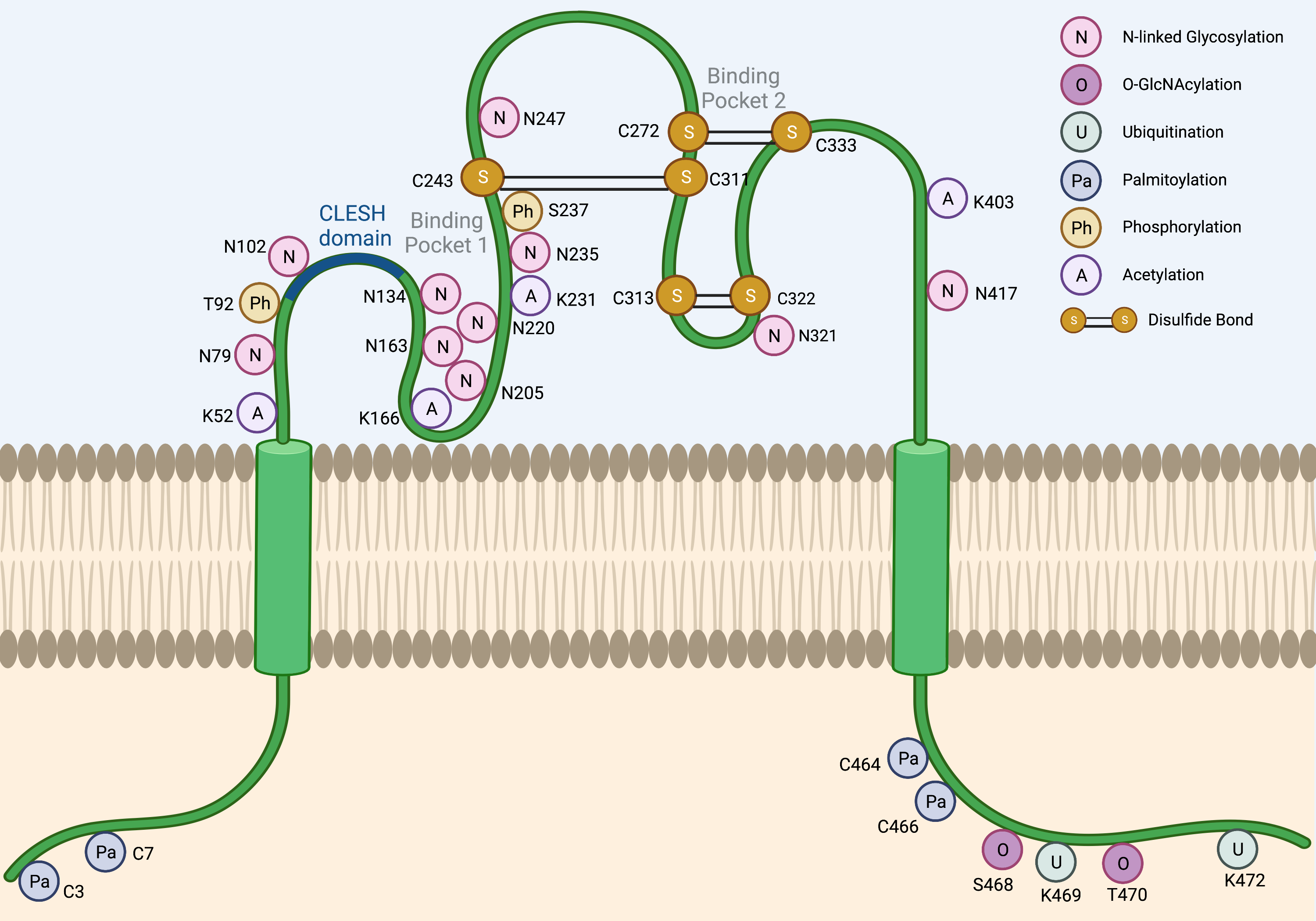
Figure 1. Post-translational modifications of CD36. Several post-translational modifications have been described for CD36 including N-linked glycosylation (N), O-GlcNAcylation (O), ubiquitination (U), palmitoylation (Pa), phosphorylation (Ph), acetylation (A), and disulfide bonds (S-S). The CLESH domain (93–120) of CD36 plays a critical role in the interaction with thrombospondins. Lys164 is known to be the binding site of FAs, oxLDLs, and the CD36 inhibitor sulfo-N-succinimidyl oleate (SSO).
2. Glycosylation
Although the predicted size of the CD36 protein is 53 kDa based on its amino acid composition, the observed molecular weight across tissues is often ~88 kDa due to glycosylation. N-linked glycosylation is a critical post-translational modification required for the maturation, stabilization, and proper trafficking of most cell surface proteins. CD36 harbors 10 potential N-linked glycosylation sites, which are all located in its extracellular domain. A mutagenesis study has demonstrated that when all 10 asparagine residues are mutated to glutamines, CD36 is retained in the cytoplasm and is unable to localize to the cell surface [2]. Notably, no single residue alone is solely responsible for determining the trafficking fate of CD36. In fact, the glycosylation pattern required for cell surface localization seems to be flexible due to a degree of functional redundancy in these residues. For instance, the restoration of either the first seven glycosylation sites (Asn79, Asn102, Asn134, Asn163, Asn205, Asn220, and Asn235) or the last three glycosylation sites alone (Asn247, Asn321, and Asn417) was sufficient to partially rescue CD36 cell surface localization [2]. However, the greatest degree of rescue could be seen when all 10 glycosylation sites were restored [2]. Importantly, N-linked glycosylation of CD36 does not appear to affect ligand binding [2].
O-linked-N-acetylglucosaminylation (O-GlcNAcylation) is another form of glycosylation that involves the addition of the sugar moiety N-acetylglucosamine (GlcNAc) with serine or threonine residues of proteins. It was recently reported that CD36 harbors two potential O-GlcNAcylation sites on Ser468 and Thr470 [3]. O-GlcNAc Transferase (OGT) and O-GlcNAcase (OGA) are the endogenous enzymes that catalyze the addition and removal of O-GlcNAc from proteins, respectively. The pharmacologic inhibition of OGA was recently shown to activate the NF-κB pathway in gastric cancer cells to markedly induce CD36 transcription, as well as increase the O-GlcNAcylation of CD36 protein [3]. The O-GlcNAcylation of CD36 subsequently enhanced cellular FA uptake and promoted metastasis of gastric cancer models [3]. Interestingly, the high expression level of both CD36 and OGT predicted a worse clinical outcome in gastric cancer patients [3]. Although the O-GlcNAcylation of CD36 has also been reported in cardiomyocytes [4], it remains unclear how prominent role O-GlcNAcylation plays to regulate CD36 functions in other cell types and cancers.
3. Ubiquitination
Lys469 and Lys472 are the only lysine residues within the CD36 intracellular domain, and both have been previously identified as critical poly-ubiquitination sites that target CD36 protein for proteasomal degradation [5]. In myoblast cells, FA binding induces CD36 poly-ubiquitination, thereby destabilizing the protein and reducing cellular FA uptake [5]. Interestingly, insulin exhibits the opposite effects in this model and stabilized CD36 to further enhance FA uptake [5]. However, the relevance of these studies in the setting of cancer is unclear since dietary FA supplementation has been shown to increase the CD36 expression level, which then promotes tumor growth and metastasis in xenograft models of oral squamous cell carcinoma [6], cervical [7], and gastric cancers [3]. Moreover, co-culture studies have revealed that omental adipocytes can induce the expression of CD36 in ovarian cancer cells to facilitate the uptake of adipocyte-derived FAs into cancer cells, promoting tumor growth and enhancing metastasis [8]. Therefore, CD36 levels in cancer cells may not be primarily regulated via FA-induced degradation.
At present, Parkin is the predominant E3 ubiquitin ligase known to promote the degradation of the CD36 protein. In mice, hepatic levels of Parkin were demonstrated to be elevated by a high-fat diet, which resulted in stabilizing CD36 protein via mono-ubiquitination [9]. However, a recent study reported that Parkin knockout mice exhibited no difference in CD36 protein levels in the brain [10]. On the other hand, it was demonstrated that the deletion of the master regulator of energy homeostasis, AMPK, promoted Parkin-mediated poly-ubiquitination and the degradation of CD36 in intestinal epithelial cells [11]. Therefore, the role of the Parkin-mediated regulation of CD36 appears to be largely context- and tissue-specific. The action of E3 ubiquitin ligases is counteracted by deubiquitinating enzymes, deubiquitinases, which stabilize the substrate proteins by removing ubiquitin. Three deubiquitinases are known to stabilize CD36 protein in macrophages: USP10 [12], USP14 [13], and UCHL1 [14]. All in all, whether Parkin, USP10, USP14, or UCHL1 plays a role in the regulation of CD36 protein stability in cancer cells remains poorly understood.
4. Palmitoylation
CD36 contains four intracellular cysteine residues, which are all known palmitoylation sites (Cys3, Cys7, Cys464, and Cys466) [15]. Interestingly, the maturation of palmitoylation-deficient CD36 mutant protein was markedly slower as compared to that of the wildtype, CD36 [16]. Additionally, the mutant CD36 also exhibited a shorter protein half-life, as well as a defect in incorporation into lipid rafts, which impairs its ability to import oxLDLs [16]. Strikingly, another study has also demonstrated that the loss of each of the four palmitoylation sites is sufficient to abrogate insulin or the AMPK-activation-induced cell surface localization of CD36, suggesting that all four palmitoylation sites are critical for this process [17]. Recently, the palmitoyl acyltransferases, DHHC4 and DHHC5, were identified to play indispensable roles in CD36 maturation [18]. DHHC4 palmitoylates synthesized CD36 in the Golgi, and the silencing of DHHC4 expression alone blocked nearly all CD36 cell surface localization. In addition, DHHC5 functions at the cell surface and helps stabilize CD36 by preventing depalmitoylation [18]. Lastly, two recent studies have independently reported that CD36 acylation/deacylation and glycosylation/deglycosylation are critical steps that govern both the uptake of extracellular FAs, as well as the trafficking of CD36 to/from lipid droplets during lipolysis in adipocytes [19][20]. Therefore, the palmitoylation of CD36 plays a critical role in its protein functions as an FA/oxLDL transporter.
5. Other Post-Translational Modifications
While glycosylation and palmitoylation are critical for CD36 maturation, phosphorylation plays a role in the regulation of ligand binding and FA uptake, as well as signal transduction activity. CD36 has two known phosphorylation sites in its extracellular domain, Thr92 and Ser237, which are phosphorylated using PKC and PKA, respectively [21][22]. It was shown that Thr92 phosphorylation using PKC can take place intracellularly before trafficking to the cell surface [23]. Under basal conditions in melanoma cells, CD36 is predominantly non-phosphorylated and presumed to be active [23]. The treatment of these cells with a PKC activator resulted in an increase in the rates of CD36 phosphorylation and the concomitant reduction of TSP-1 peptide binding, which consequently abolished the ligand-induced recruitment of SRC to CD36 [23]. Likewise, it was shown that treatment with a PKA inhibitor peptide reduced the rate of palmitate uptake into the platelets [24]. In contrast to Thr92 phosphorylation using PKC, however, the role and the mechanism of Ser237 phosphorylation using PKA remain poorly characterized to date. It has been shown that PKA can function at the cell surface and phosphorylate the extracellular domain of CD36 [22]. However, this does not rule out the possibility of the intracellular phosphorylation of CD36 using PKA prior to trafficking to the plasma membrane. Importantly, an earlier study reported that PKA could phosphorylate CD36, but less efficiently than PKC [21].
The acetylation of human CD36 has been described for Lys52, Lys16, Lys231, and Lys403 [25]. The acetylation of Lys166 may impact the FA uptake activity of CD36 since acetylated Lys166 and FA-bound Lys164 have not been found on the same protein in mass spectrometry experiments [25]. However, the functional significance and prevalence of CD36 acetylation in cancer remain uncharacterized.
The extracellular domain of CD36 harbors three disulfide bonds that are critical for its structure. Interestingly, the Cys272-Cys333 disulfide bond in CD36 was shown to be cleavable using hydrogen sulfide (H2S), and the loss of this specific motif enhanced FA uptake and metastasis of gastric cancer cells [26]. The translatability of this finding to other cancer types and models requires further study.
6. Transcriptional Regulation of CD36
In addition to the regulation of CD36 maturation, trafficking, activity, and stability via post-translational modifications, CD36 is also regulated at the transcriptional level. Many transcription factors have been shown to regulate CD36 expression, including PPARγ [27][28][29], LXR [28], PXR [28], C/EBPα [30], STAT3 [31][32], HIF1α [3][8], SOX2 [3], and NF-κB [3][8]. Some of these transcription factors are known to play a role in human diseases, including cancer. For instance, the activation of NF-κB could trigger lipid metabolic rewiring in ovarian cancer cells, thereby activating a transcriptional program that leads to increased CD36 expression, which in turn can be critical for ovarian cancer survival and metastasis [8]. Interestingly, Protein Kinase D1 (PKD1) is known to be a potent activator of NF-κB [33][34], and it is often aberrantly activated in cancer [35][36]. PKD1 signaling has been demonstrated to mediate the induction of CD36 to facilitate the maintenance of a stem-like phenotype in pancreatic neuroendocrine tumors [36]. The transcriptional upregulation of CD36 has been well documented in other cancers. However, transcription factors responsible for CD36 upregulation in cancer are largely unknown.
References
- Tandon, N.N.; Lipsky, R.H.; Burgess, W.H.; Jamieson, G.A. Isolation and characterization of platelet glycoprotein IV (CD36). J. Biol. Chem. 1989, 264, 7570–7575.
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The human scavenger receptor CD36: Glycosylation status and its role in trafficking and function. J. Biol. Chem. 2009, 284, 16277–16288.
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019, 9, 5359.
- Nabeebaccus, A.A.; Zoccarato, A.; Hafstad, A.D.; Santos, C.X.; Aasum, E.; Brewer, A.C.; Zhang, M.; Beretta, M.; Yin, X.; West, J.A.; et al. Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight 2017, 2, e96184.
- Smith, J.; Su, X.; El-Maghrabi, R.; Stahl, P.D.; Abumrad, N.A. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: Effects on fatty acid uptake. J. Biol. Chem. 2008, 283, 13578–13585.
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenuer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting Metastasis-Initiating Cells through the Fatty Acid Receptor CD36. Nature 2017, 541, 41–45.
- Tandon, N.N.; Kralisz, U.; Jamieson, G.A. Identification of glycoprotein IV (CD36) as a primary receptor for platelet-collagen adhesion. J. Biol. Chem. 1989, 264, 7576–7583.
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301.
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 2037–2047.
- Tang, W.; Thundyil, J.; Lim, G.G.; Tng, T.J..; Yeow, S.Q.; Chai, C.; Yao, T.P.; Lim, K.L. Parkin regulates neuronal lipid homeostasis through SREBP2-lipoprotein lipase pathway—Implications for Parkinson’s disease. Hum. Mol. Genet. 2022, 32, 1466–1482.
- Wu, W.; Wang, S.; Liu, Q.; Shan, T.; Wang, X.; Feng, J.; Wang, Y. AMPK facilitates intestinal long-chain fatty acid uptake by manipulating CD36 expression and translocation. FASEB J. 2020, 34, 4852–4869.
- Xia, X.; Hu, T.; He, J.; Xu, Q.; Yu, C.; Liu, X.; Shao, Z.; Liao, Y.; Huang, H.; Liu, N. USP10 deletion inhibits macrophage-derived foam cell formation and cellular-oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging 2020, 12, 22892.
- Zhang, F.; Xia, X.; Chai, R.; Xu, R.; Xu, Q.; Liu, M.; Chen, X.; Liu, B.; Liu, S.; Liu, N. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J. Cell. Mol. Med. 2020, 24, 3292–3302.
- Xia, X.; Xu, Q.; Liu, M.; Chen, X.; Liu, X.; He, J.; Hu, T.; Yu, C.; Huang, H.; Liu, S. Deubiquitination of CD36 by UCHL1 promotes foam cell formation. Cell Death Dis. 2020, 11, 636.
- Tao, N.; Wagner, S.J.; Lublin, D.M. CD36 is palmitoylated on both N-and C-terminal cytoplasmic tails. J. Biol. Chem. 1996, 13, 22315–22320.
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta Mol Cell. Res. 2010, 1803, 1298–1307.
- van Oort, M.M.; Drost, R.; Janβen, L.; Van Doorn, J.M.; Kerver, J.; Van der Horst, D.J.; Liuken, J.J.F.P.; Rodenburg, K.C.W. Each of the four intracellular cysteines of CD36 is essential for insulin-or AMP-activated protein kinase-induced CD36 translocation. Arch. Physiol. Biochem. 2014, 120, 40–49.
- Wang, J.; Hao, J.W.; Wang, X.; Guo, H.; Sun, H.H.; Lai, X.Y.; Liu, L.Y.; Zhu, M.; Wang, H.Y.; Li, Y.F. DHHC4 and DHHC5 facilitate fatty acid uptake by palmitoylating and targeting CD36 to the plasma membrane. Cell Rep. 2019, 26, 209–221.
- Daquinag, A.C.; Gao, Z.; Fussell, C.; Immaraj, L.; Pasqualini, R.; Arap, W.; Akimzhanov, A.M.; Febbraio, M.; Kolonin, M.G. Fatty acid mobilization from adipose tissue is mediated by CD36 posttranslational modifications and intracellular trafficking. JCI Insight 2021, 6, e147057.
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765.
- Asch, A.S.; Liu, I.; Briccetti, F.M.; Barnwell, J.W.; Kwakye-Berko, F.; Dokun, A.; Goldberger, J.; Pernambuco, M. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 1993, 262, 1436–1440.
- Hatmi, M.; Gavaret, J.M.; Elalamy, I.; Vargaftig, B.B.; Jacquemin, C. Evidence for cAMP-dependent platelet ectoprotein kinase activity that phosphorylates platelet glycoprotein IV (CD36). J. Biol. Chem. 1996, 271, 24776–24780.
- Chu, L.Y.; Silverstein, R.L. CD36 ectodomain phosphorylation blocks thrombospondin-1 binding: Structure-function relationships and regulation by protein kinase C. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 760–767.
- Guthmann, F.; Maehl, P.; Preiss, J.; Kolleck, I.; Rüstow, B. Ectoprotein kinase-mediated phosphorylation of FAT/CD36 regulates palmitate uptake by human platelets. Cell. Mol. Life Sci. 2002, 59, 1999–2003.
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-Succinimidyl Oleate (SSO) Inhibits Fatty Acid Uptake and Signaling for Intracellular Calcium via Binding CD36 Lysine 164: SSO Also Inhibits Oxidized Low Density Lipoprotein Uptake by Macrophages. J. Biol. Chem. 2013, 288, 15547–15555.
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 2019, 45, 108–123.
- Liang, Y.; Han, H.; Liu, L.; Duan, Y.; Yang, X.; Ma, C.; Zhu, Y.; Han, J.; Li, X.; Chen, Y. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis 2018, 7, 98.
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology 2008, 134, 556–567.
- Ghodsian, N.; Yeandle, A.; Hock, B.D.; Gieseg, S.P. CD36 down regulation by the macrophage antioxidant 7, 8-dihydroneopterin through modulation of PPAR-γ activity. Free Radic. Res. 2022, 56, 366–377.
- Qiao, L.; Zou, C.; Shao, P.; Schaack, J.; Johnson, P.F.; Shao, J. Transcriptional regulation of fatty acid translocase/CD36 expression by CCAAT/enhancer-binding protein α. J. Biol. Chem. 2008, 283, 8788–8795.
- Gyamfi, J.; Yeo, J.H.; Kwon, D.; Min, B.S.; Cha, Y.J.; Koo, J.S.; Jeong, J.; Lee, J.; Choi, J. Interaction between CD36 and FABP4 Modulates Adipocyte-Induced Fatty Acid Import and Metabolism in Breast Cancer. NPJ Breast Cancer 2021, 7, 129.
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.M.; Yang, Y.K. Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the Cd36/Stat3/Nf-Κb signaling axis. Nutrients 2018, 10, 772.
- Yuan, J.; Lugea, A.; Zheng, L.; Gukovsky, I.; Edderkaoui, M.; Rozengurt, E.; Pandol, S.J. Protein kinase D1 mediates NF-κB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1190–G1201.
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 2003, 22, 109–120.
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A Novel Small-Molecule Inhibitor of Protein Kinase D Blocks Pancreatic Cancer Growth In vitro and In vivo. Mol. Cancer Ther. 2010, 9, 1136–1146.
- Guo, Y.; Jiang, Y.; Rose, J.B.; Nagaraju, G.P.; Jaskula-Sztul, R.; Hjelmeland, A.B.; Beck, A.W.; Chen, H.; Ren, B. Protein Kinase D1 Signaling in Cancer Stem Cells with Epithelial-Mesenchymal Plasticity. Cells 2022, 11, 3885.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
15 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No